




Bone Tumors
An appropriate and effective study for bone tumors by the orthopedic tumor surgeon with his team (orthopedic tumor radiologist, orthopedic tumor pathologist, vascular surgeon, microsurgeon with sufficient reconstruction experience, radiation oncologist and medical oncologist) is essential for the patient's treatment to be adequate, otherwise the patient will be treated incorrectly. Diagnosis or delayed diagnosis will result in serious disabilities and death.
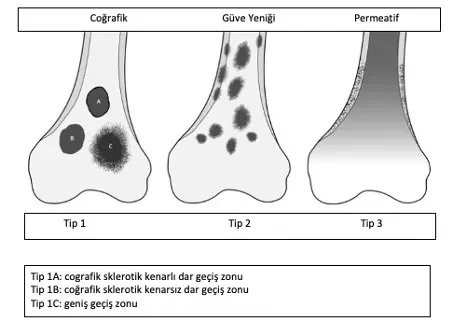
Diagnosis begins with taking a detailed history of the patient's complaint, a detailed examination and a classic x-ray. Benign tumors are generally seen as lesions that do not cause any complaints, have geographical features, sclerotic edges, and do not cause bone destruction or periosteal reaction.
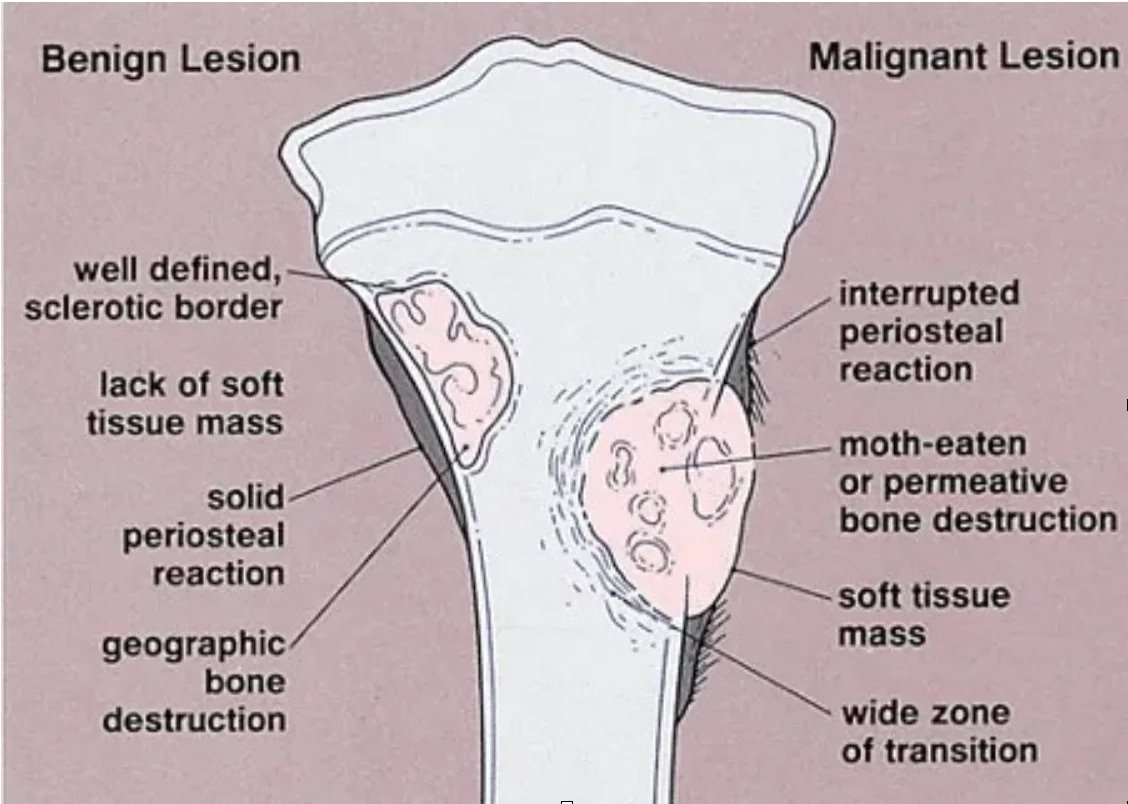
Malignant tumors, on the other hand, cause regional pain and show lytic, permeative growth and ill-defined borders with intact bone on radiographs. Bone membrane destruction and/or periosteal reaction (Picture 2) may be observed.
At this stage, a classical orthopedic doctor should refer his patient to a tumor orthopedic specialist. Advanced imaging and biopsy are methods to be decided by your tumor doctor.
Clinic of Bone Tumors
The clinic is very variable. Lesions in patients may occur accidentally or with symptoms such as pain and swelling. For example, fibrous dysplasia or osteochondromatosis may occur with bone curvature in patients. Fractures, soft tissue masses and swelling may occur along with the tumor in the bone. Painful lesions are generally seen in aggressive (benign but aggressive) and potentially malignant tumors. Since the clinical spectrum of benign and malignant tumors is very wide, diagnosis should be made by a physician experienced in tumor surgery. Incorrect and inadequate diagnoses and treatments will expose a patient who could be managed with simple follow-up or treatment to unnecessary, expensive and damaging tests and treatments. Likewise, inadequate diagnosis and treatment of aggressive and malignant tumors may result in loss of limbs or loss of life.
Clinic in Benign Tumors
There may be moderate pain that is relieved with painkillers. Pain occurs slowly and may be associated with activity or trauma. For example, night pain that responds to painkillers is typical in osteoid osteoma. Fractures that occur with a movement that would not normally break the bone are called pathological fractures. In some benign lesions, pathological fractures may develop with single or repeated traumas. IF THE LESION IS DETECTED ACCIDENTALLY WHILE THE PATIENT HAS NO PAIN OR SYMPTOMS, THEN THEY ARE MOST LIKELY BENEFICIAL.
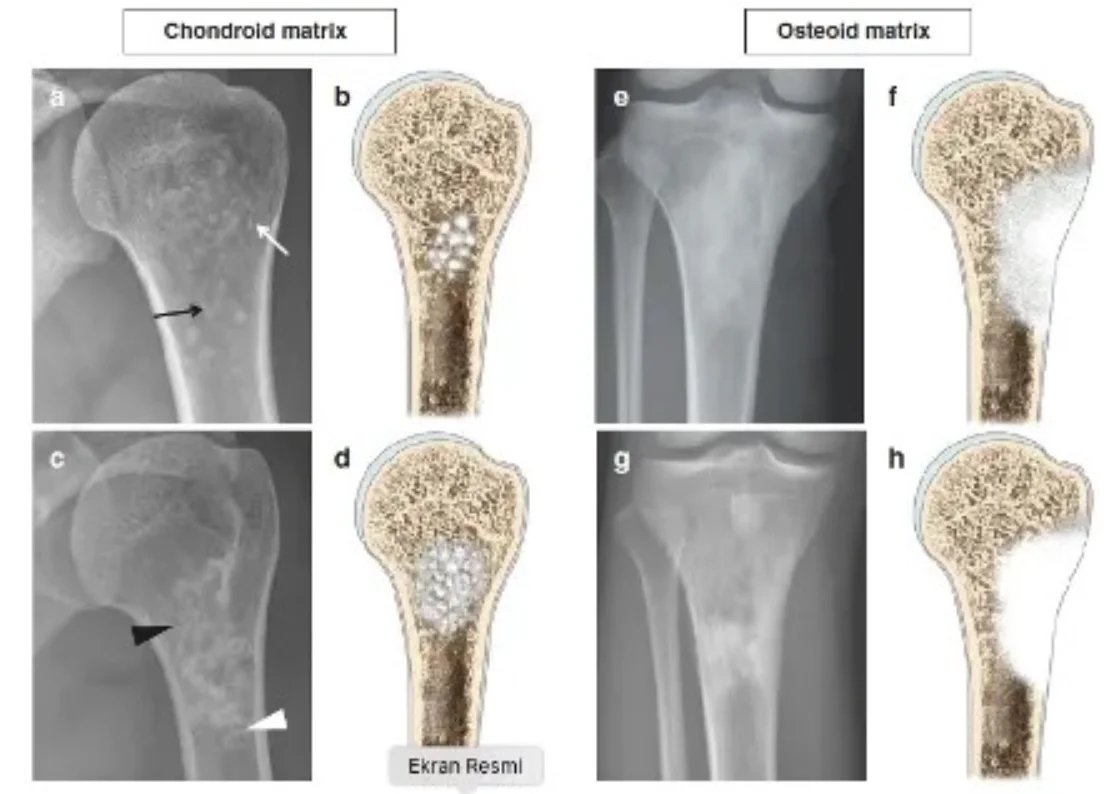
An experienced tumor surgeon will understand whether the lesion is good or requires a biopsy based on the patient's age, location of the lesion, and x-ray. Radiological examination should include the entire bone. Benign tumors are typically geographical, have narrow transition zones and sclerotic edges (Figure 1). Endosteal thinning may occur, but cortex destruction is rare. Another helpful criterion in diagnosis is the matrix of the lesion.
MRI shows the soft tissue component, contrast material retention, bone marrow edema and signal characteristics of the lesion. Scintigraphy is effective in poliostatic fibrous dysplasia, multiple enchondromatosis and histiocytosis to see multiple bone involvements. A cold scintigraphy does not indicate that the lesion is aggressive or non-malignant (e.g. multiple myeloma, renal cell carcinoma metastasis). Degenerative and overuse lesions in lesions adjacent to the joint are also included in the differential diagnosis.
If there is cortex destruction, permeative growth and periosteal reaction, further examination is essential. If the diagnosis is not clear despite CT and MRI, a biopsy is required. After the diagnosis of benign bone tumor is made, observation and follow-up can be done at 3-6 month intervals to ensure radiological stability. Surgical intervention will be required to stop bone destruction, detect pathological fractures or strengthen lesions that are about to break, and prevent deformities.
Clinic in Malignant Tumors
The clinic is very variable. Lesions in patients may occur accidentally or with symptoms such as pain and swelling. For example, patients with fibrous dysplasia or osteochondromatosis usually show signs of bone curvature. The pain is often severe and painkillers are not enough. There is a dull and deep pain unrelated to activity or rest. If a pathological fracture occurs, pain occurs suddenly. Numbness in the feet and legs, weakness, bowel and bladder dysfunction are seen in spine and sacrum tumors. There may be an accompanying swelling. Fatigue, weakness and fever may occur. Increased alkaline phosphatase, elevated calcium and anemia may be seen in the laboratory. Family history of cancer and sarcoma is important in the history. For example, retinablastoma, Li Fraumeni syndrome and Rothmund-Thompson syndromes have a predisposing effect on bone tumors such as osteosarcoma. It has been reported that multiple enchondromatosis lesions may worsen. In addition, sarcoma may develop in Paget patients, history of radiotherapy, chronic osteomyelitis and bone infarction. The biopsy should be performed by an orthopedic tumor surgeon because the biopsy should be done as closed as possible and this requires experience. In addition, the biopsy site should be chosen so as not to contaminate important vessels and nerves. The biopsy site must be on the line preferred by the tumor surgeon in the actual tumor surgery. Otherwise, long and heavy surgeries such as serious muscle and tissue losses, dangerous vascular transplants, nerve losses and skin transplants will be required. This may increase limb loss and shorten life expectancy. If a malignant bone tumor is diagnosed, staging should be done. For this, PET imaging must be performed quickly. In patients over 40 years of age, the lesion is usually metastatic carcinoma, multiple myeloma or lymphoma. In addition, the breast in women and the prostate in men should be evaluated and protein electrophoresis should be performed in serum and urine. Treatment may require a multidisciplinary approach, including radiation and medical oncologists, under the leadership of an orthopedic tumor surgeon. Fractures, soft tissue masses and swelling may occur along with the tumor in the bone. Painful lesions are generally seen in aggressive (benign but aggressive) and potentially malignant tumors. Since the clinical spectrum of benign and malignant tumors is very wide, diagnosis should be made by a physician experienced in tumor surgery. Incorrect and inadequate diagnoses and treatments will expose a patient who could be managed with simple follow-up or treatment to unnecessary, expensive and damaging tests and treatments. Likewise, inadequate diagnosis and treatment of aggressive and malignant tumors may result in loss of limbs or loss of life.
Soft Tissue Tumors
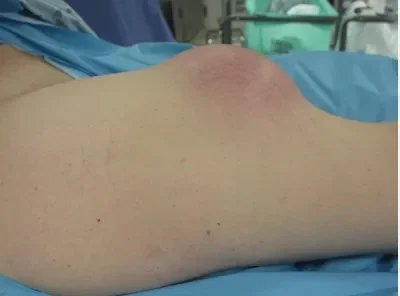
Soft tissue tumors are more common than bone tumors and are often benign. The diagnosis process begins with a detailed history and examination. X-ray, MRI, ultrasound, CT and PET are used for advanced examination. These tools are also used to evaluate treatment response. Benign ones are 100 times more common than malignant ones. The location, size and consistency of the mass are important in the history and examination. Its depth is checked relative to the fascia.
Questions to ask:
- When was the mass noticed?
- Is there any increase in size?
- What is the growth rate?
- Are there any reductions in size?
- Is there an accompanying mass?
- Are there pain, skin redness, regional lymph node swelling?
- Is there fever, chills, night sweats?
In addition, questions should be asked about issues such as trauma, blood thinner use, previous cancer, and world travel.
A SMALL, SOFT, SUPERFICIAL AND STABLE MASS FOR YEARS IS VERY PROBABLY BENEFICIAL.
LARGE, HARD, DEEP AND GROWING MASSES HAVE A HIGH POSSIBILITY OF BEING MALICIOUS.
Hematomas may occur with mild trauma in people using blood thinners, but these hematomas may mask an underlying cancer.
The presence of ossification in the mass helps in differential diagnosis. For example, in a benign mass due to trauma called myositis ossificans, ossification occurs in the periphery (peripheral), while in soft tissue osteosarcoma (malignant) it occurs in the center. Calcification has a more disorganized appearance than ossification. Phleboliths can be seen in vascular malformations and dystrophic calcifications can be seen in some lipomas. If this dystrophic calcification appears aggressive, the diagnosis suggests synovial sarcoma, which is extremely malignant. If multiple uniform calcifications are around the joint, a benign tumor called synovial chondromatosis should come to mind. An aggressive periosteal reaction or cortical destruction seen in the surrounding bone tissue adjacent to the mass on radiography should suggest a high probability that the mass is bad. High signal on T2 on MRI, mass being larger than 3 cm, being under the fascia, peripheral edema, hemorrhage within the mass, heterogeneity on T1, tumor necrosis, bone or vascular nerve invasion, peripheral, nodular, heterogeneous contrast enhancement indicate that the mass is most likely bad. .
Thanks to the use of medication in MRI (contrast MRI), hematomas, cystic-solid distinction, cystic-necrotic areas within the mass are determined, and thus the most appropriate biopsy site is distinguished. However, both cystic masses and tumors rich in myxoid structure can shine on liquid-sensitive sequences. These masses can only be distinguished by medicated MRI. In cystic masses, the drug keeps the drug peripherally, while solid masses hold solid contrast.
If radiotherapy and/or chemotherapy was administered before surgery, it is necessary to examine the tumor with a new MRI before surgery. In this way, the treatment response can be understood. Sometimes, despite these treatments, the mass may grow, one of the reasons for this is that the mass begins to die and bleed from within, which can be revealed by medicated MRI.
Sarcomas
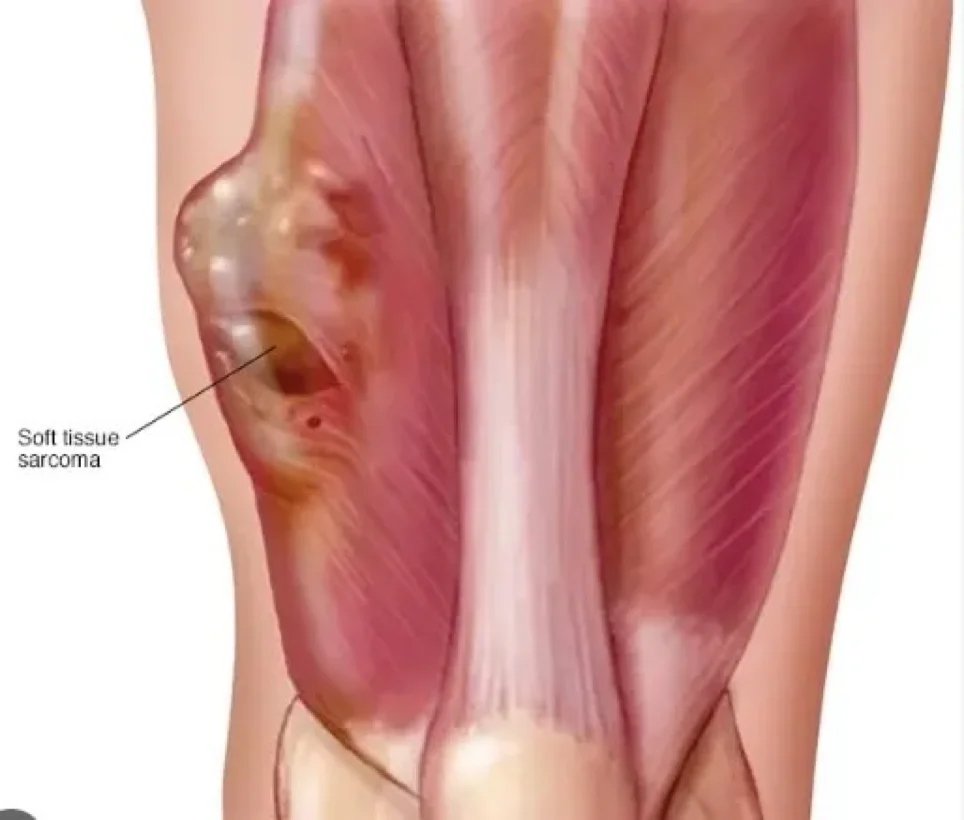
Sarcoma is a malignant tumor, a type of cancer arising from transformed cells of mesenchymal (connective tissue) origin. Connective tissue is a broad term that includes bone, cartilage, fat, vascular or hematopoietic tissues, and sarcomas can occur in any of these tissue types. As a result, there are many subtypes of sarcoma, classified according to the specific tissue and cell type from which the tumor originates. Sarcomas are primary connective tissue tumors, meaning they arise from connective tissues.
This is in contrast to secondary (or “metastatic”) connective tissue tumors, which occur when a cancer from elsewhere in the body (such as the lungs, breast tissue, or prostate) spreads to the connective tissue. The word sarcoma is derived from the Greek word σάρκωμα sarōma “fleshy stool or substance”, itself from σάρξ sarx meaning “flesh”.
Classification
Sarcomas are typically divided into two main groups: bone sarcomas and soft tissue sarcomas, each of which has multiple subtypes[2]. In the United States, the American Joint Committee on Cancer (AJCC) publishes guidelines classifying sarcoma subtypes.
These subtypes are as follows:
Subtypes of bone sarcoma:
- Osteosarcomachondrosarcoma
- Poorly differentiated round/spindle cell tumors (including Ewing sarcoma) hemangioendothelioma
- angiosarcoma
- Fibrosarcoma/myofibrosarcomachordoma
- Adamantima
Others:
- Liposarcomaleiomyosarcoma
- Malignant peripheral nerve sheath tumor
- Rhabdomyosarcoma
- Synovial sarcoma
- Malignant solitary fibrous tumor
Subtypes of soft tissue sarcoma:
- Liposarcoma (includes the following types: atypical lipomatous tumor/well-differentiated liposarcoma, dedifferentiated liposarcoma, myxoid liposarcoma, pleomorphic liposarcoma, and myxoid pleomorphic liposarcoma
- Dermatofibrosarcoma protuberans (includes pigmented varieties)
- Dermatofibrosarcoma protuberans, fibrosarcomatosis
- Giant cell fibroblastoma
- Malignant solitary fibrous tumor
- Inflammatory myofibroblastic tumor
- Low grade myofibroblastic sarcoma
- Fibrosarcoma (includes adult and sclerosing epithelioid varieties)
- Myxofibrosarcoma (formerly myxoid malignant fibrous histiocytoma)
- Low grade fibromyxoid sarcoma
- Giant cell tumor of soft tissues (leiomyosarcoma)
- Malignant glomus tumor
- Rhabdomyosarcoma (includes: embryonal, alveolar, pleomorphic, and spindle cell/sclerosing)
- Hemangioendothelioma (includes: retiform, pseudomyogenic, and epithelioid)
- Soft tissue angiosarcoma
- Extraskeletal osteosarcoma
- Gastrointestinal stromal tumor, malignant (GIST)Malignant peripheral nerve sheath tumor (includes epithelioid variety)
- Malignant Triton tumor
- Malignant granular cell tumor
- Malignant ossifying fibromyxoid tumor
- Stromal sarcoma not otherwise specified
- Myoepithelial carcinoma
- Malignant phosphaturic mesenchymal tumor
- Synovial sarcoma (includes: spindle cell, biphasic, and not otherwise specified)
- epithelioid sarcoma
- Alveolar soft part sarcoma
- Clear cell sarcoma of soft tissue
- Extraskeletal myxoid chondrosarcomaExtraskeletal
- Ewing sarcomaInterdigitating dendritic cell sarcoma
- Desmoplastic small round cell tumor
- Extrarenal rhabdoid tumor
- Perivascular epithelioid cell tumor, not otherwise specified
- intimal sarcoma
- Undifferentiated spindle cell sarcoma
- Undifferentiated pleomorphic sarcoma
- Undifferentiated round cell sarcoma
- Undifferentiated epithelioid sarcoma
- Undifferentiated sarcoma, not otherwise specified.
Sarcoma Symptoms:
Symptoms of bone sarcomas typically include bone pain, especially at night, and swelling around the tumor site.
The symptoms of soft tissue sarcomas vary, but they usually present as hard, painless lumps or nodules. Gastrointestinal stromal tumors (a subtype of soft tissue sarcoma) are usually asymptomatic but may be associated with vague complaints such as abdominal pain, a feeling of fullness, or other signs of intestinal obstruction.
Causes and risk factors:
The cause of most bone sarcomas is unknown, but several factors are associated with an increased risk of developing bone sarcoma. Previous exposure to ionizing radiation (such as prior radiation therapy) is one such risk factor. Exposure to alkylating agents, such as those found in some cancer chemotherapeutic drugs, also increases the risk of bone sarcoma. Certain inherited genetic syndromes, including Li-Fraumeni syndrome, hereditary RB1 gene mutations, and Paget bone disease, are associated with an increased risk of developing bone sarcomas.
Most soft tissue sarcomas are caused by what doctors call “sporadic” (or random) genetic mutations in the affected person's cells. However, there are certain risk factors associated with an increased risk of developing soft tissue sarcoma. Previous exposure to ionizing radiation is one such risk factor. Exposure to Vinyl chloride (such as fumes encountered in the production of Polyvinyl chloride (PVC)), Arsenic, and Thorotrast are associated with an increased risk of angiosarcoma. Lymphedema, such as that resulting from some breast cancer treatments, is also a risk factor for developing angiosarcoma. As with bone sarcomas, certain inherited genetic syndromes, including Li-Fraumeni syndrome, familial adenomatous polyposis, neurofibromatosis type 1, and hereditary RB1 gene mutations, are also associated with an increased risk of developing soft tissue sarcomas. Kaposi's sarcoma is caused by Kaposi's sarcoma-associated herpes virus (HHV-8).
Mechanisms:
Although the exact molecular changes that result in sarcoma are not always known, some types of sarcoma are associated with certain genetic mutations.
Examples include:
Most cases of Ewing sarcoma are associated with a chromosomal translocation in which part of chromosome 11 joins part of chromosome 22.[2] This causes the EWS gene to fuse with other genes, including the FLI1 gene in 90% of Ewing cases and the ERG gene in 5-10% of cases. These fusions cause abnormal proteins to be produced, but it is not known exactly how these abnormal proteins cause cancer.
Dermatofibrosarcoma protuberans is often associated with a chromosomal translocation in which the COL1A1 gene is fused to the PDGFRB gene. This results in overactive PDGF signaling, which is thought to promote cell division and ultimately lead to tumor development.
Inflammatory myofibroblastic tumor is often associated with rearrangements of the ALK gene and sometimes rearrangements of the HMGA2 gene.
Giant cell tumor of soft tissue is often associated with a chromosomal translocation between chromosome 1 and chromosome 2, where the CSF1 gene is fused with the COL6A3 gene. This causes increased production of CSF1 protein, which is thought to play a role in cancer development.
Many liposarcomas are associated with duplication of part of chromosome 12, resulting in extra copies of known cancer-promoting genes (“oncogenes”), such as the CDK4 gene, MDM2 gene, and HMGA2 gene.
Diagnosis:
Bone sarcomas
Diagnosis of bone sarcomas begins with a detailed history and physical examination, which can reveal characteristic signs and symptoms (see Signs and Symptoms above). Although some bone sarcomas (such as osteosarcoma) may be associated with elevated alkaline phosphatase levels and others (such as Ewing Sarcoma) with an elevated erythrocyte sedimentation rate, laboratory studies are not particularly useful in diagnosis. Importantly, however, none of these laboratory findings are specific to bone sarcomas; This means that elevations in these laboratory values are associated with many other conditions besides sarcoma and therefore cannot be relied upon to definitively diagnose sarcoma. Imaging studies are critical in diagnosis, and most clinicians initially order a plain radiograph (x-ray). Other imaging studies commonly used in diagnosis include magnetic resonance imaging (MRI) studies and radioisotope bone scans. Although an important tool for staging, computed tomography (CT) imaging is not typically used to diagnose most types of bone sarcomas. Definitive diagnosis requires biopsy of the tumor and careful examination of the biopsy sample by an experienced pathologist.
Soft tissue sarcomas
Diagnosis of soft tissue sarcomas begins with a comprehensive history and physical examination. Imaging studies may include CT or MRI, but CT tends to be preferred for soft tissue sarcomas located in the thorax, abdomen, or retroperitoneum. Positron emission tomography (PET) can also be useful in diagnosis, but its most common use is for staging. As with bone sarcomas, definitive diagnosis requires biopsy and evaluation of histology by a trained pathologist.
Staging:
In general, cancer staging refers to how advanced a cancer is and is often based on factors such as the size of the tumor and whether it has spread to other parts of the body. Staging is important because stage affects prognosis (likely outcome) as well as the types of treatments that are likely to be effective against the cancer. In staging sarcomas, it is checked whether the tumor has grown into surrounding tissues (local invasion), spread to lymph nodes (nodal metastases), or invaded other tissues or organs in the body (distant metastases).
The most common imaging tools used to stage bone sarcomas are MRI or CT to evaluate the primary tumor, contrast-enhanced CT of the chest to evaluate whether the cancer has spread (i.e., metastasized) to the lungs, and radioisotope bone scan to evaluate bone metastases. Staging for soft tissue sarcomas typically includes imaging of the primary tumor with MRI or CT to determine tumor size, as well as contrast-enhanced chest CT to evaluate metastatic tumors in the lungs.
Grade:
Like some other cancers, sarcomas are assigned a grade (low, medium, or high) based on the appearance of the tumor cells under the microscope. In general, grade refers to how aggressive the cancer is and how likely it is to spread (“metastasize”) to other parts of the body. Low-grade sarcomas have a better prognosis than high-grade sarcomas and are usually treated surgically. Intermediate and high-grade sarcomas are more often treated with a combination of surgery, chemotherapy, or radiation therapy. Because higher-grade tumors are more likely to metastasize (invade and spread to regional and distant sites), they are treated more aggressively. Knowing that many sarcomas are sensitive to chemotherapy has significantly improved patient survival. For example, in the pre-chemotherapy era, long-term survival in pediatric patients with localized osteosarcoma was only 20%, but it has now increased to 60-70%.
Treatment
The most common treatment for most sarcomas that have not spread to other parts of the body is surgery. We now perform limb-sparing surgery, as opposed to amputation, to save patients' limbs in at least 90% of extremity (arm or leg) sarcoma cases. Additional treatments, including chemotherapy, radiation therapy (also called “radiotherapy”), and proton therapy, may be administered before surgery (called “neoadjuvant” chemotherapy or radiotherapy) or after surgery (called “adjuvant” chemotherapy or radiotherapy). The use of neoadjuvant or adjuvant chemotherapy and radiotherapy has significantly improved the prognosis of many sarcoma patients. Treatment can be a long and arduous process, taking about a year for many patients.
Prognosis
Factors affecting prognosis
The AJCC has identified several factors that affect the prognosis of bone sarcomas:
Tumor size:Larger tumors tend to have a worse prognosis than smaller tumors.
Tumor spread to surrounding tissues: Tumors that have spread locally to surrounding tissues tend to have a worse prognosis than tumors that have not spread beyond their site of origin.
Stage and presence of metastases: Tumors that have spread (“metastasized”) to the lymph nodes (rare for bone sarcomas) or other organs or tissues (for example, the lungs) have a worse prognosis than tumors that have not spread.
Tumor grade:high-grade tumors (grades 2 and 3) tend to have a worse prognosis than low-grade (grade 1) tumors.
Skeletal location: Tumors originating from the spine or pelvic bones tend to have a worse prognosis than tumors originating from the bones of the arms or legs.
Factors affecting prognosis for soft tissue sarcomas other than GIST include:
Grade: The AJCC recommends the use of a grading system called the French Federation of Cancer Centers Sarcoma Group (FNCLCC) Grade for soft tissue sarcomas, where higher-grade tumors have a worse prognosis than lower-grade tumors. p>
Key factor affecting prognosis for GISTs:
Mitotic rate: mitotic rate refers to the fraction of cells actively dividing within the tumor; GISTs with a high mitotic rate have a worse prognosis than GISTs with a low mitotic rate.
Result Data
According to data published by the US National Cancer Institute (NCI), the overall 5-year survival for bone sarcomas is 66.9%. The American Cancer Society (ACS) estimates that 1,660 people in the United States will die from bone sarcomas in 2019, accounting for 0.3% of all cancer deaths. The average age of death is 61, but death can occur in any age group. Thus, 12.3% of bone sarcoma deaths occur in people under the age of 20, 13.8% in people between the ages of 20-34, 5.5% in people between the ages of 35-44, and 9.3% in people between the ages of 45-54. 13.5% in people aged 55-64, 16.2% in people aged 65-74, 16.4% in people aged 75-84 and 13.1% in people aged 85 and up. It is seen in people above.
The overall 5-year survival for soft tissue sarcomas (regardless of stage) is 64.5%, but survival is affected by many factors, including stage. Therefore, the 5-year survival is 80.8% for soft tissue sarcomas that have not spread beyond the primary tumor (“localized” tumors), 58.0% for soft tissue sarcomas that have spread only to nearby lymph nodes, and 16% for soft tissue sarcomas with distant organ spread. is .4. The ACS estimates that 5,270 people will die from soft tissue sarcoma in 2019, accounting for 0.9% of all cancer deaths.
Epidemiology
Sarcomas are extremely rare. The risk of a previously healthy person being diagnosed with a new bone cancer is less than 0.001% (1 in 100,000), while the risk of being diagnosed with a new soft tissue sarcoma is between 0.0014 and 0.005%. The American Cancer Society estimates there will be 3,500 new cases of bone sarcoma and 12,750 new cases of soft tissue sarcoma in the United States in 2019. Considering that the estimated total number of new cancer diagnoses (all cancer types) is 1,762,450, this suggests that bone sarcomas represent only 0.2% of all new cancer diagnoses (making them the 30th most common type of cancer). Soft tissue sarcomas represented only 0.7% of all new cancer diagnoses in the United States in 2019 (making them the 22nd most common type of cancer).
Sarcomas affect people of all ages. Approximately 50% of bone sarcomas and 20% of soft tissue sarcomas are diagnosed in people under 35 years of age. Some sarcomas, such as leiomyosarcoma, chondrosarcoma, and gastrointestinal stromal tumor (GIST), are more common in adults than in children. Most high-grade bone sarcomas, including Ewing sarcoma and osteosarcoma, are much more common in children and young adults.
Treatment:
Treatment of sarcoma often requires chemotherapy, especially when the sarcoma has spread or “metastasized,” but current chemotherapeutic drugs are associated with significant toxicities and are not fully effective at killing cancer cells. Therefore, research to identify new drugs to treat sarcoma is being conducted as of 2019. One possibility is to use cancer immunotherapy (e.g., immune checkpoint inhibitors such as anti-PD1, anti-PDL1, and anti-CTLA4 agents) to treat sarcomas. This is not yet an established treatment tool. Other strategies such as small molecule targeted therapy, biological agents (e.g., small interfering RNA molecules), and nanoparticle-focused therapy are also being investigated.
Dr Sameer Rastogi et al. has shown long-lasting responses to immunotherapy in several sarcomas (UPS & ASPS).
Research continues to understand the specific genetic and molecular factors that cause sarcoma to develop. This could allow new targeted therapies to be designed and allow doctors to more accurately predict a patient's prognosis.
The presence of the H3-B3 immunoregulatory checkpoint receptor on tumor cells provides the opportunity for clinical trial testing of new drugs and targeted agents and immunotherapies in development.
Awareness:
In the United States, July is widely recognized as Sarcoma Awareness Month. The UK has Sarcoma Awareness Week in July, led by the bone and soft tissue cancer charity Sarcoma UK.
American YouTuber Technoblade was diagnosed with sarcoma in August 2021 and died due to his disease in June 2022 after the cancer metastasized. He raised over $500,000 in a charity campaign. Many YouTubers raised awareness and donated to charities like the Sarcoma Foundation of America after Technoblade was diagnosed and passed away.
Biopsy
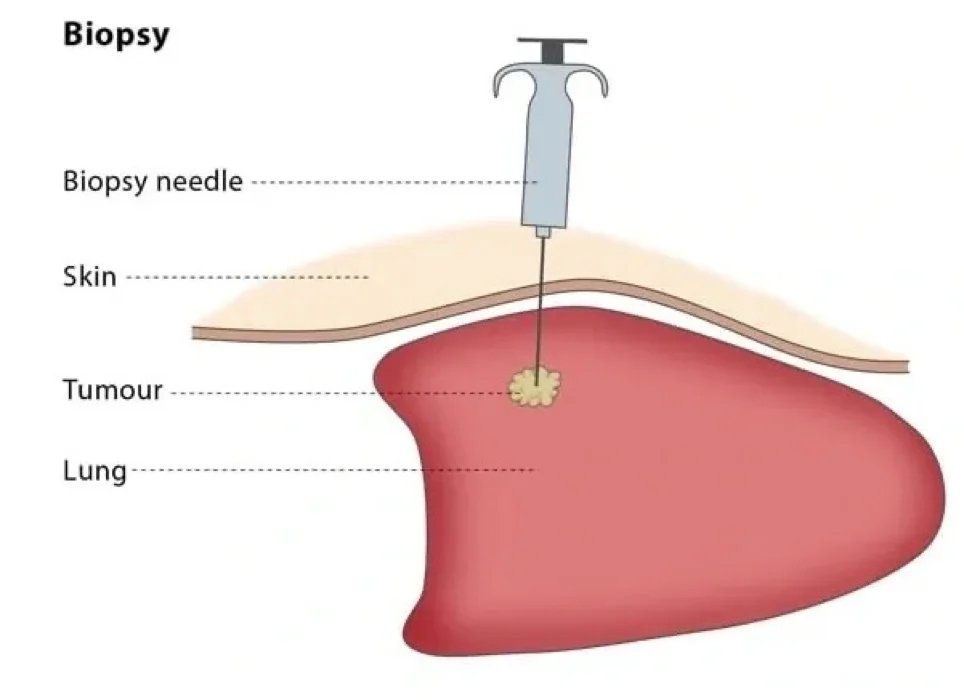
One of the most important stages in the diagnosis and treatment of musculoskeletal system tumors is biopsy. A well-planned and performed biopsy will minimize injuries that may occur due to the disease. It is a common belief among the public that a knife cannot be applied to a tumor. Actually, I think this is true. For this reason, we rarely perform open biopsies.
In recent years, when we evaluated small samples taken without opening the tumor using special needles with experienced pathology instructors, we started to make diagnoses with high accuracy rates. Since we take biopsies with a needle and closed method, the spread of tumor cells is minimal. The main principle of tumor surgery is to remove the mass without seeing the tumor or entering the tumor, leaving some healthy tissue around. In other words, the tumor should not really be stabbed.
Biopsy Principles:
- Cancer surgery cannot be performed without a cancer diagnosis.
- Clinical and radiological evaluation must be complete before biopsy
- Although benign masses are much more common than malignant masses, the diagnosis of cancer should always be considered.
- Closed biopsy is sufficient in most cases and should be preferred.
- If an open biopsy is to be performed, it must be performed by the surgeon who will perform the surgery.
- The method called excisional biopsy (considering the entire mass as biopsy material without taking any part and performing wide resection) should only be performed in special cases.
- Sarcoma diagnosis and treatment is team work. It is necessary to leave the job to competent hands.
If these principles are followed, the most accurate diagnosis can be achieved with the least harm to the patient. These are:
- Lipomas (fat glands): if the lesion is homogeneous and isointense with fat tissue in all sequences, no biopsy is required
- Benign bone tumors with typical radiology (ostechondromas, non-ossifying fibromas, enchondromas, unicameral simple bone cysts). In fact, these lesions may not require further examination with high-quality x-rays.
- In addition to lipomas (soft tissue masses with typical MRI findings), schwannomas, hemangiomas, arteriovenous malformations, myxomas.
- Metastatic lesions in patients with biopsy-proven carcinoma or myeloma
I usually perform soft tissue biopsies under local anesthesia under office conditions. I perform the biopsy with a needle in about ten minutes, with minimal pain. The patient can return to work on the same day. Even though I work with one of our most experienced instructors in closed needle biopsies, diagnosis can rarely be made. On this issue, some authors advocate switching to open biopsy, while others advocate continuing closed biopsy until the diagnosis is made. Personally, if I try closed biopsy twice and still cannot be diagnosed, I prefer to switch to open biopsy.
I perform bone biopsies in the operating room under scopy imaging, preferably as a closed needle biopsy. I take biopsy samples under general anesthesia in a procedure that takes approximately 15 minutes so that the patient does not feel any pain.
If the patient is suspected of infection during imaging, blood tests or examination before the procedure, antibiotics should be discontinued, at least two weeks should be waited, and cultures should be performed for aerobic, anaerobic, acid-fast bacteria and fungi. In the past, the approach that a culture should be taken from every biopsy and a biopsy should be taken from every culture was common. However, thanks to modern imaging and laboratory methods, it has become possible to narrow down the possibility of infection in biopsy patients. Unnecessary cultures may lead to incorrect diagnosis of infection, which may expose the patient to long, tiring and expensive antibiotic treatments.
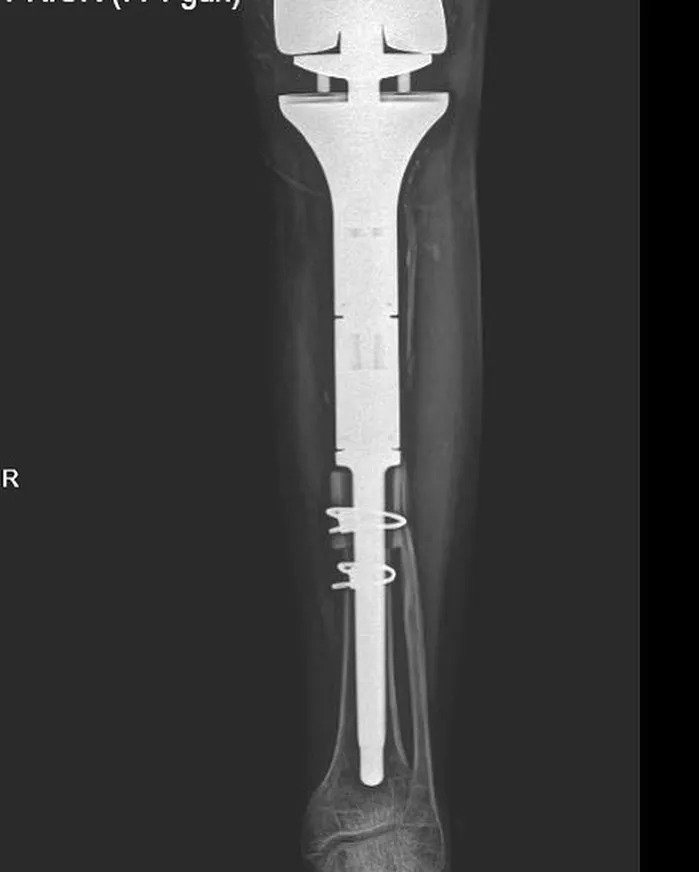
Osteosarcoma patient
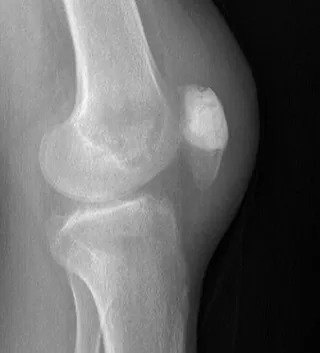
Patella Aneurysmal Bone Cyst
Extremely rare case
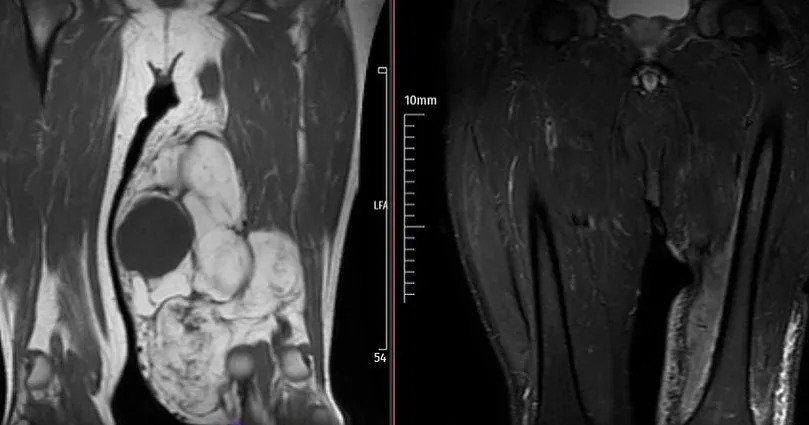
Dedifferentiated Liposarcoma Case
No recurrence with curettage and cementation
Dedifferentiated Chondrosarcoma
The residual tumor in the patient, who underwent inadequate surgery 10 years ago, had deteriorated and reached a gigantic size. Extensive resection was performed and the patient regained his health.
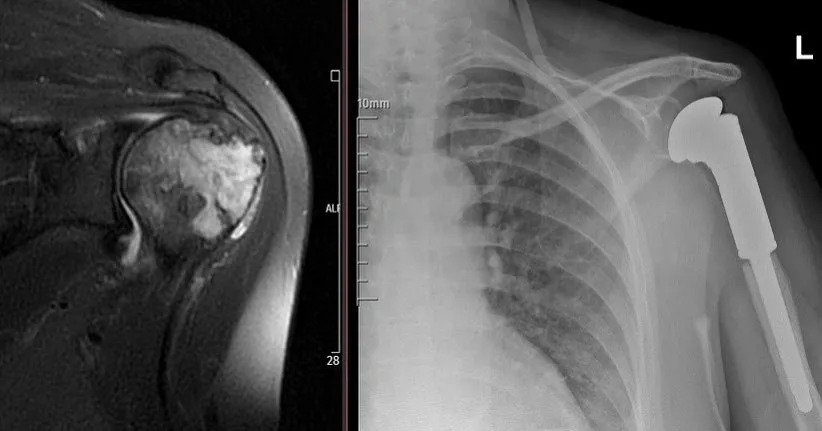
Giant Cell Bone Tumor
It is very rare and very malignant but we have not had any recurrence or metastasis for two years.
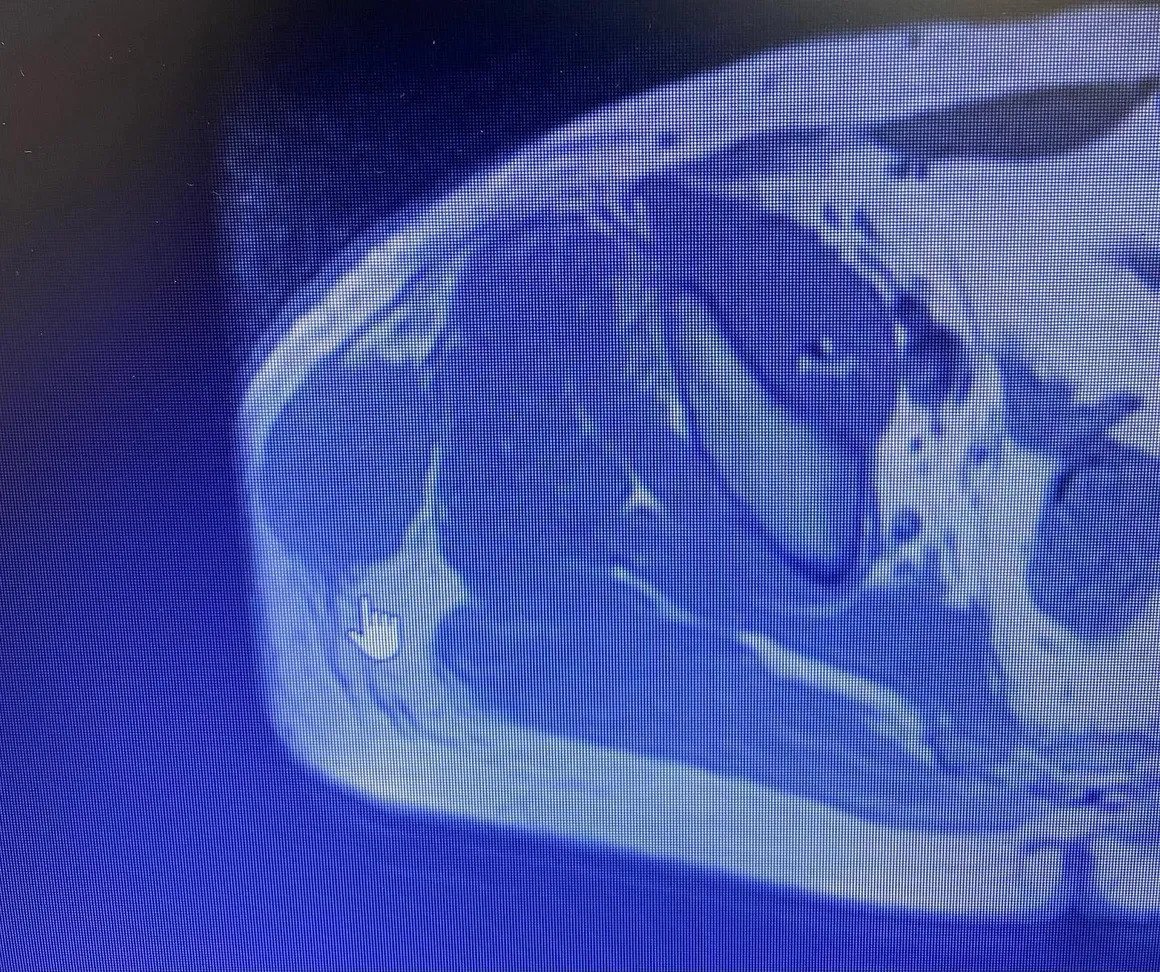
Soft tissue Ewing Sarcoma
Giant cell tumor is benign but has a devastating effect on bone
Soft tissue Ewing Sarcoma
Soft tissue Ewing sarcomas are extremely rare. The patient regained his health with chemotherapy and surgery
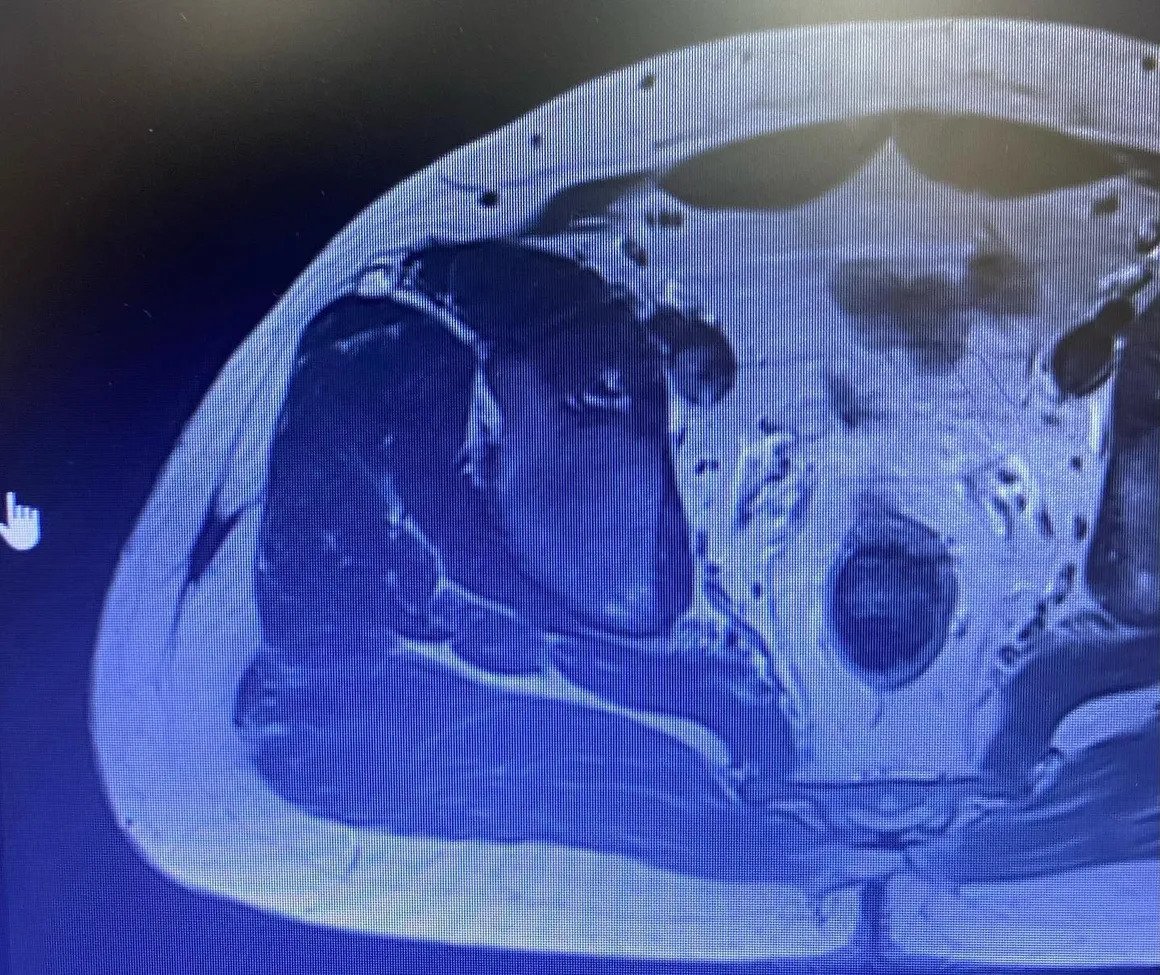
Femur Lower End Osteosarcoma
Soft tissue Ewing sarcomas are extremely rare. The patient regained his health with chemotherapy and surgery
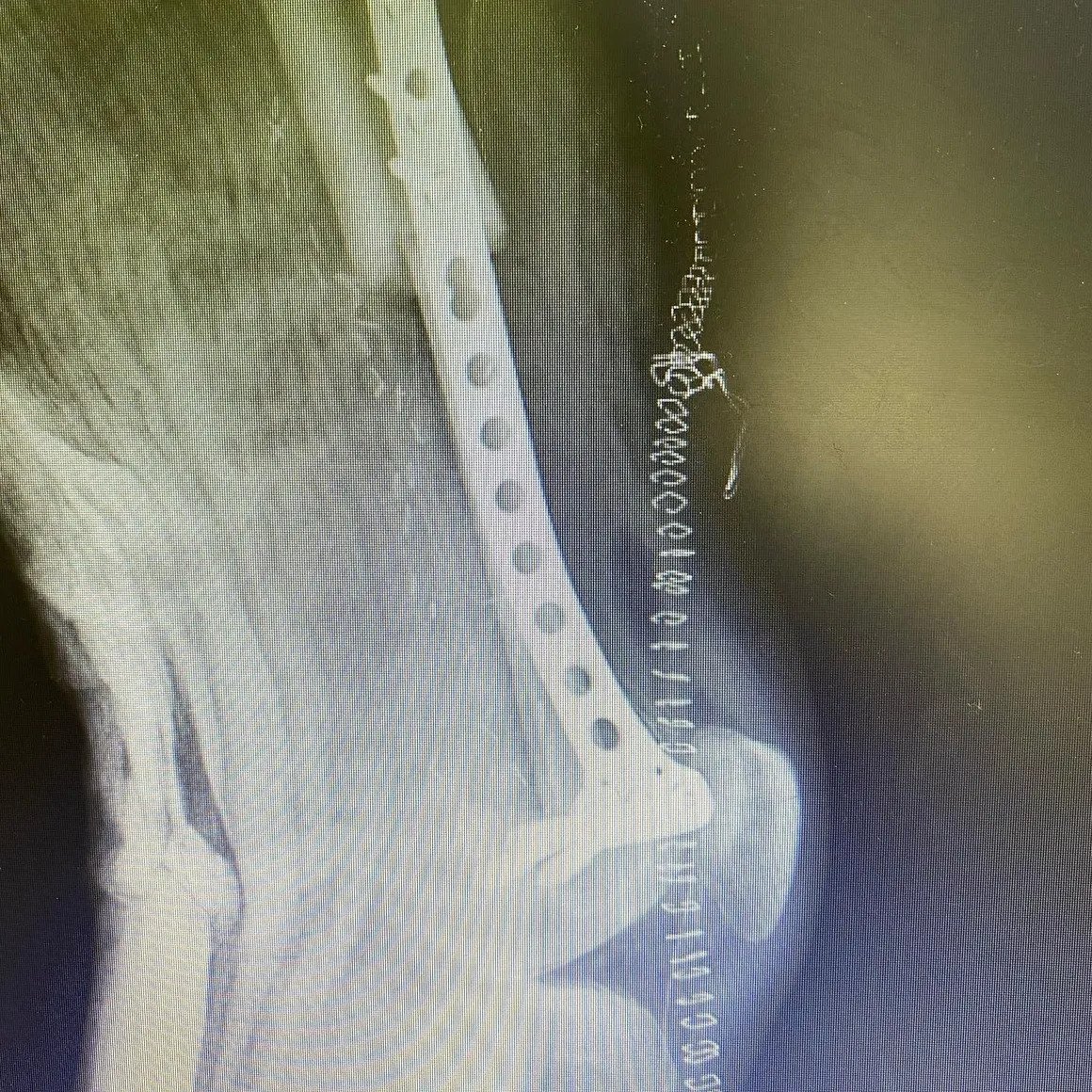
Femur Lower End Osteosarcoma
After chemotherapy, extensive resection and vascularized fibula bone transplantation saved his leg.
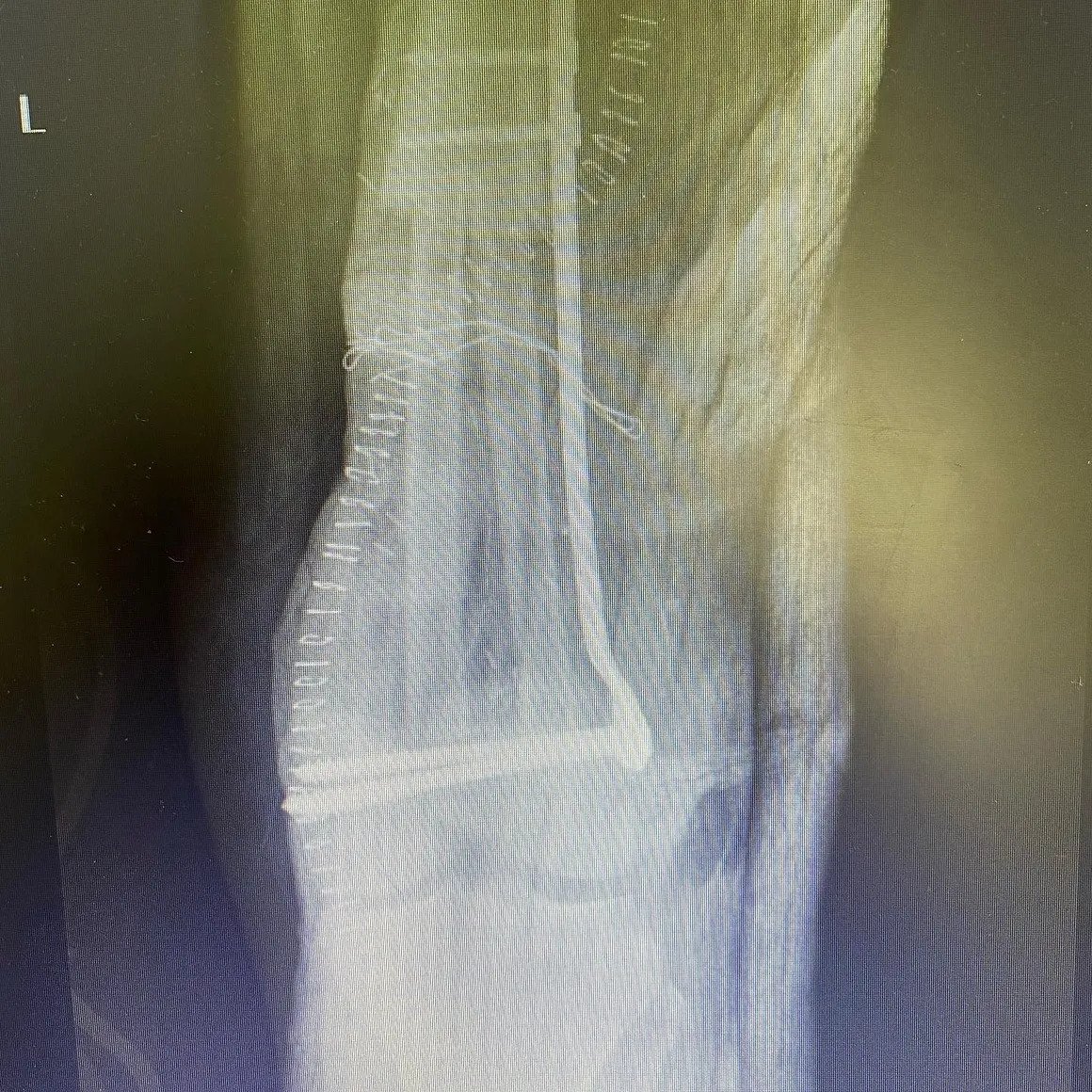
Femur Lower End Osteosarcoma
Atypical Cartilage Tumor
After chemotherapy, extensive resection and vascularized fibula bone transplantation saved his leg.

Osteoid Osteoma Case
It's not cancer, but it can turn into cancer. He got rid of the mass completely with open surgery.
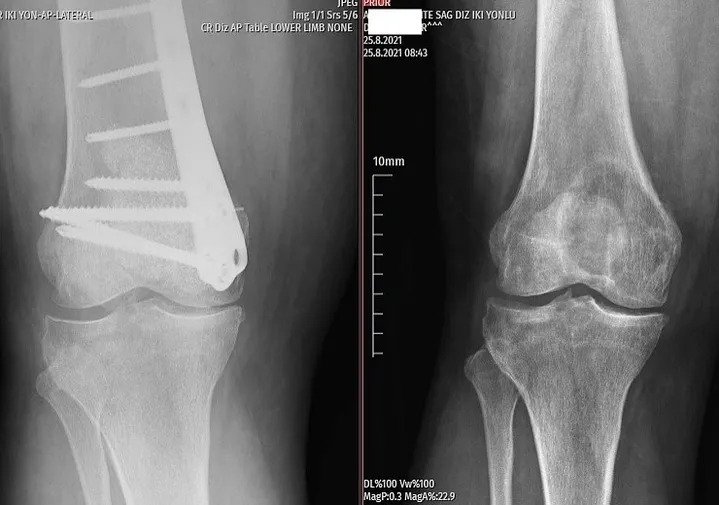
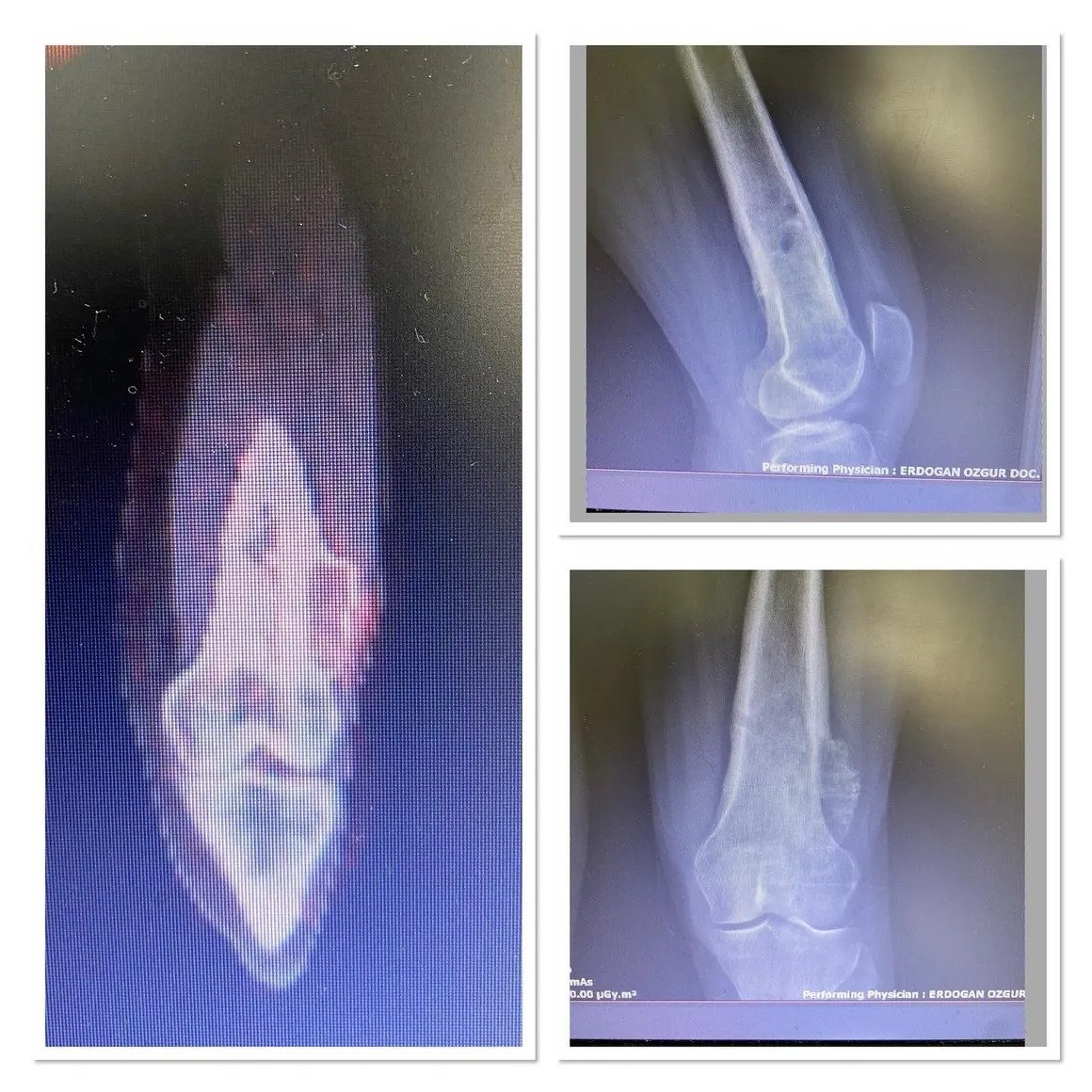
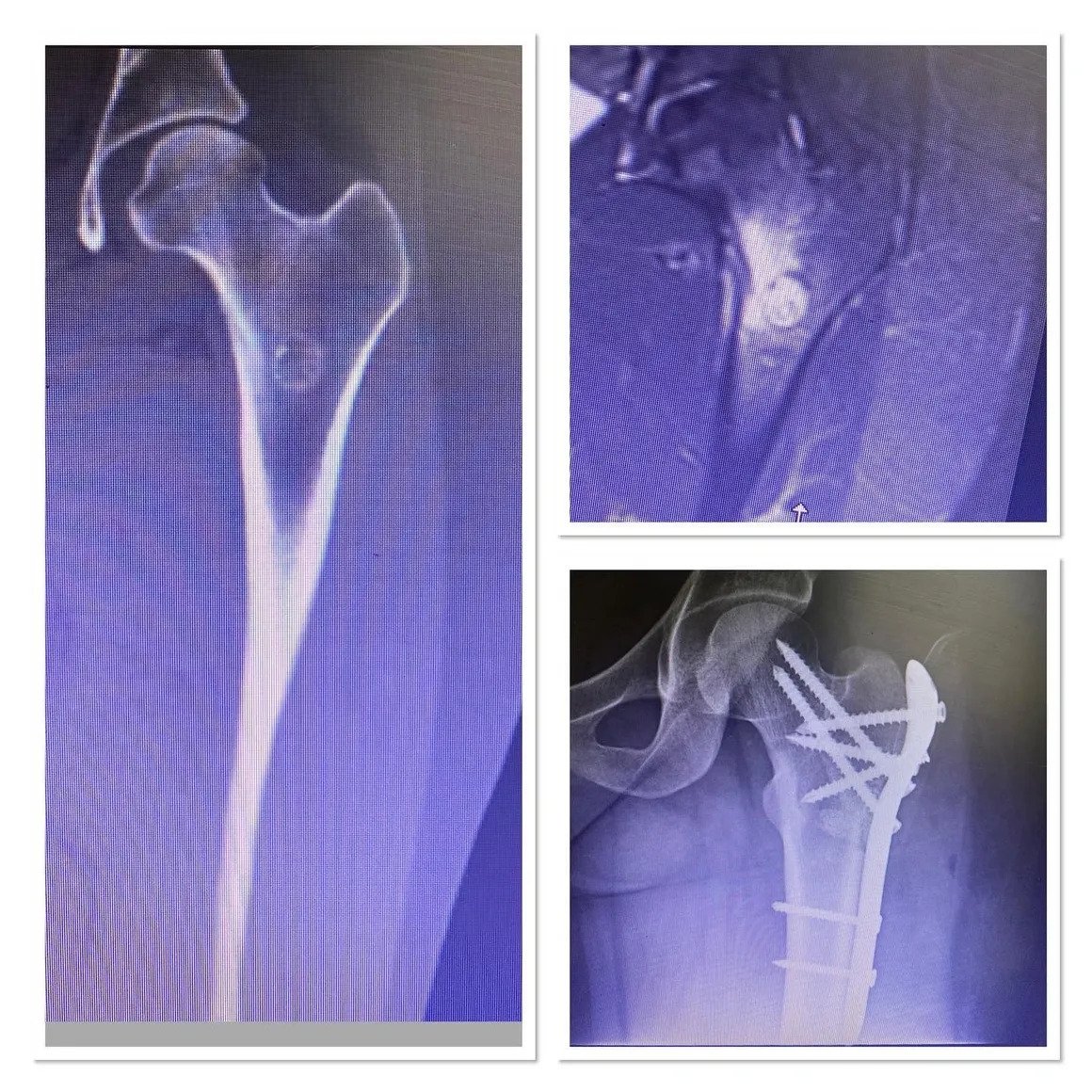
Fibula Osteosarcoma
With curettage, the pain disappeared the next day
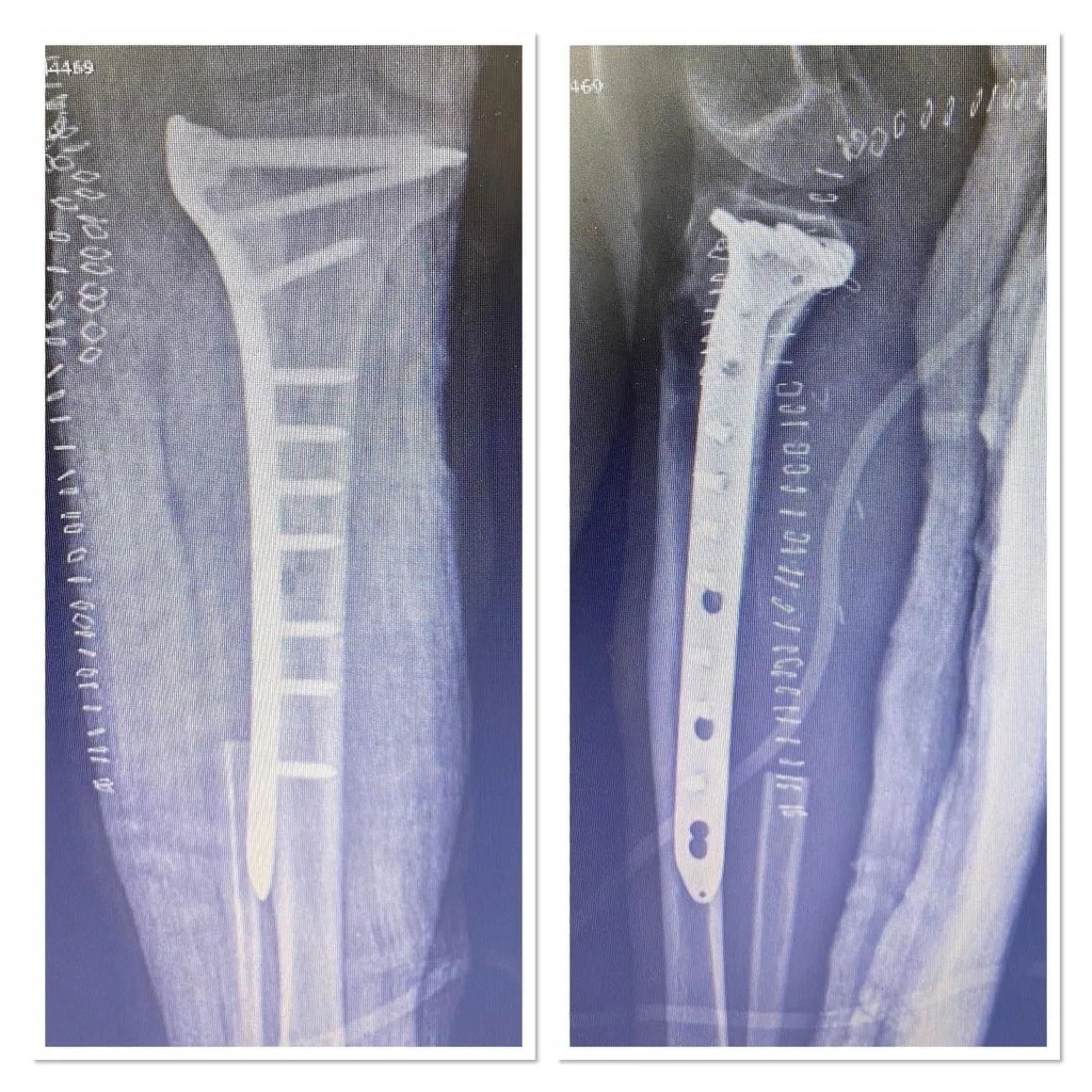
Fibula Osteosarcoma
Fibula tumors are one of the most difficult orthopedic tumor surgeries after pelvic tumors. In this patient, the tumor was extensively removed and vascular repair was performed.
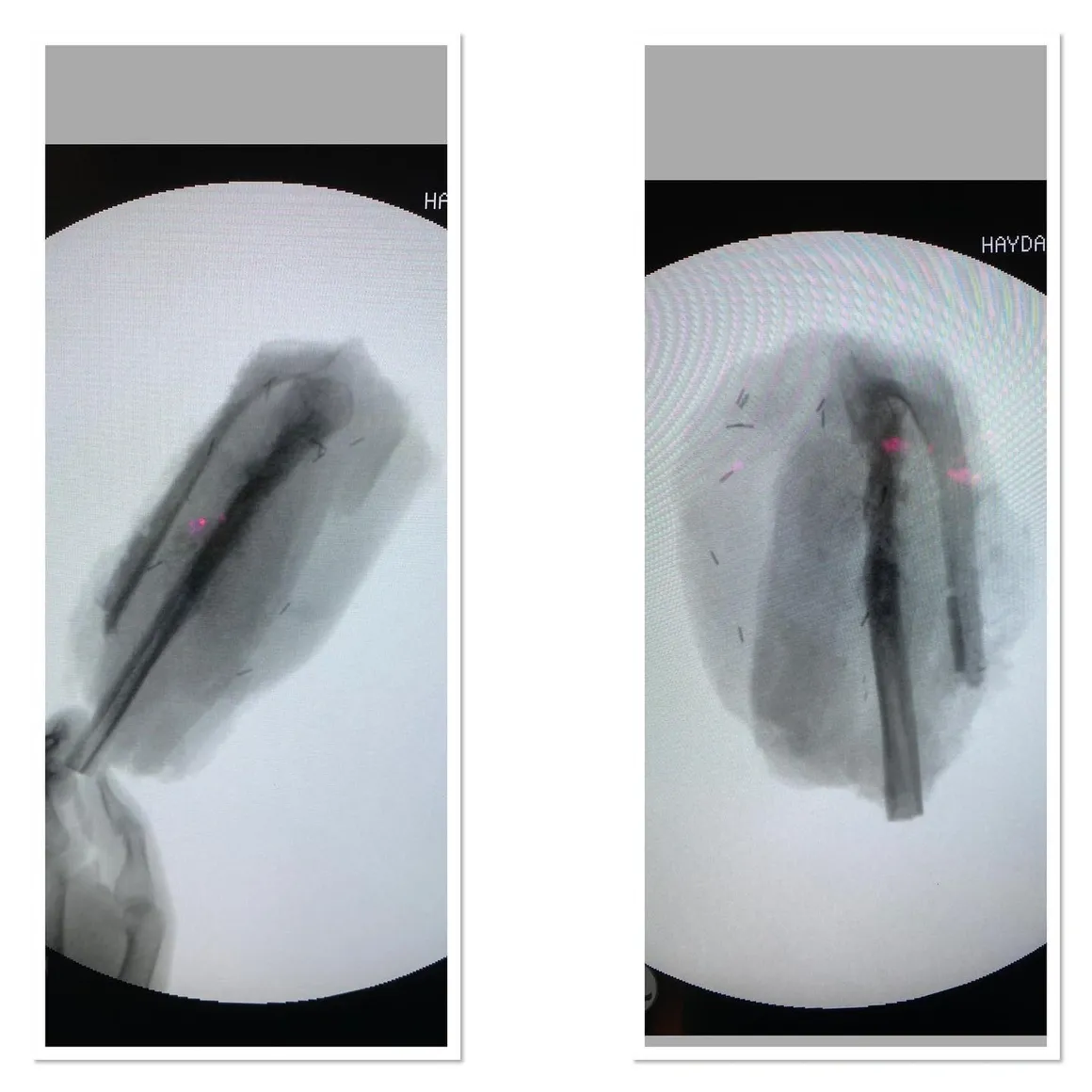
Fibula Osteosarcoma
Fibula tumors are one of the most difficult orthopedic tumor surgeries after pelvic tumors. In this patient, the tumor was extensively removed and vascular repair was performed.
