




EWING SARCCOMA TREATMENT
Ewing sarcoma is the second most common primary malignant bone tumor, affecting mostly adolescents in the second decade of life, and is a highly metastatic class of sarcoma. Despite the use of radiotherapy or surgery, an estimated 85% to 90% of patients in the past have died within a few months of metastasis. But today, due to significant progress in treating the disease with both local therapy and multi-agent adjuvant chemotherapy, the 5-year survival rate has increased from less than 20% to more than 70%. However, the recurrence rate still remains high. It must be recognized and treated promptly to prevent high morbidity and mortality. Ewing sarcoma (ES) is an aggressive tumor occurring in adolescents and young adults, accounting for 10% to 15% of all bone sarcomas.
It was first described by James Ewing in 1921. The Ewing sarcoma family includes 'classical' bone Ewing sarcoma, extraskeletal Ewing sarcoma, malignant small cell tumor of the chest wall (Askin tumor) and soft tissue-based primitive neuroectodermal tumors (PNET). The t(11;22)(q24;q12) translocation is associated with 85% of tumors and leads to the formation of EWS-FLI-1, whereas t(21;12)(22;12) and other less common translocations cause EWS-FLI Induces -1. ERG fusion accounts for the remaining 10% to 15% of cases. The most common anatomical regions include the pelvis, axial skeleton, and femur; however, it can occur in almost any bone or soft tissue. Typically, patients present with pain and swelling in the area of involvement. Although most are locally present, almost all have subclinical metastatic disease. Approximately 25% of patients with initial localized disease eventually relapse.
There is no standard treatment for recurrent and refractory ES. There is no association between ES and environmental risk factors, drug exposure, radiation history, or family history of cancer. Ewing sarcoma consists of small round cells with an increased nuclear-cytoplasmic ratio, representing the family of small round blue cell tumors of childhood (e.g., retinoblastoma, neuroblastoma, rhabdomyosarcoma, and nephroblastoma). Ewing cells have scant eosinophilic cytoplasm containing abundant glycogen, and this is usually detected by periodic acid Schiff staining. High CD99 expression has been demonstrated in more than 80% of cases. This highly sensitive immunohistochemical biomarker likely plays a key role in facilitating sustained migration of leukocytes to the endothelium; However, it has no specificity as it can also be detected in other sarcomas and lymphomas. In addition to the MIC2 gene product CD99, Ewing cells frequently express CD45, synaptophysin, chromogranin, vimentin, vimentin, keratin, desmin, neuron-specific enolase (NSE), and S-100. However, this immunohistochemistry panel is limited by lack of specificity. Molecular genetic studies using fluorescence in situ hybridization (FISH) and/or reverse transcription-polymerase chain reaction (RT-PCR) are needed to make a definitive distinction.
Ewing Sarcoma Findings: Patients with Ewing sarcoma usually present with local symptoms such as pain, stiffness, or swelling for several weeks or months. More than 50% of patients with ES have intermittent pain that worsens at night. Ewing sarcoma can occur in a wide variety of locations with different presentations. It is usually found in the diaphysis of long bones. Bone lesions or metastatic lesions in the long bone may occur as pathological fractures. The pelvic location of ES may present as back pain. The presence of systemic symptoms such as fever and weight loss often indicates metastatic disease. Approximately 20% of patients present with metastatic disease at the time of diagnosis, and more than 20% of these cases have lung or pleural involvement.
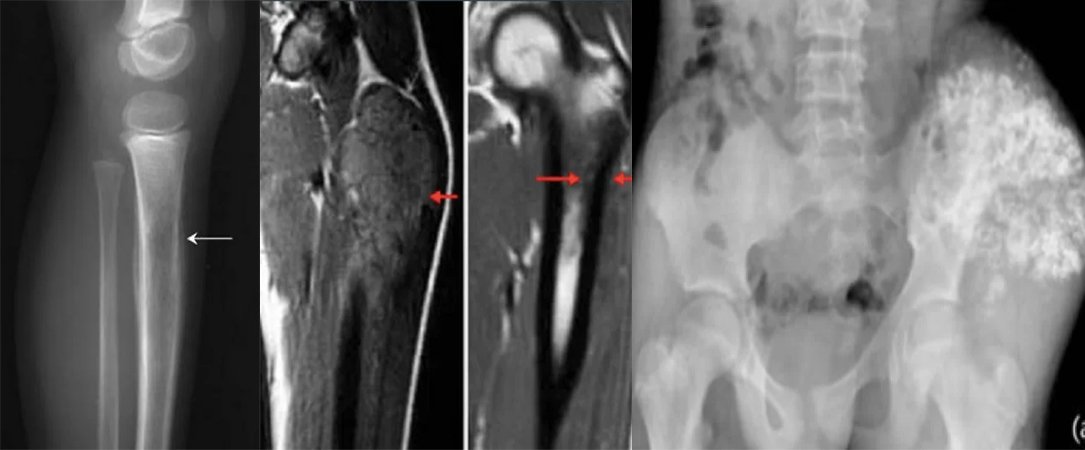
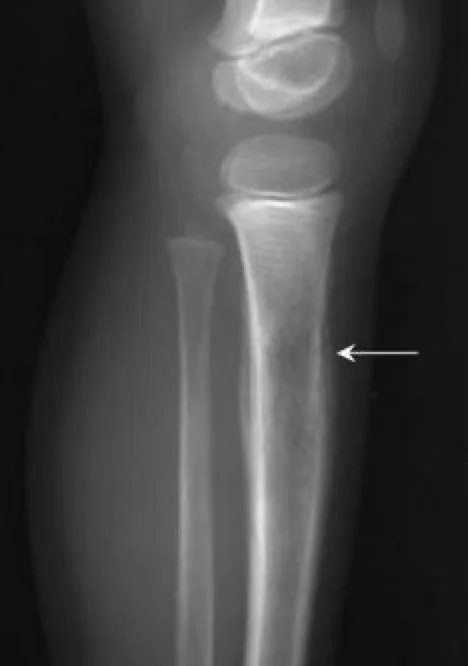
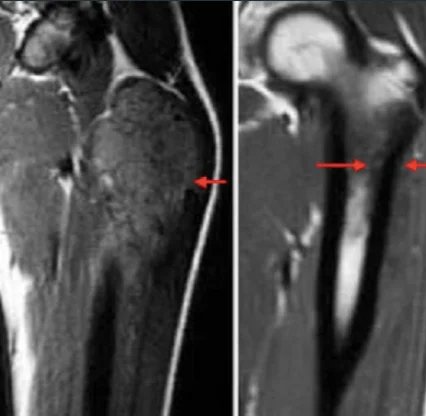
A comprehensive physical examination is important. In case of lung and pleura metastasis, the patient may present with asymmetric breath sounds, pleural symptoms or rales. In patients with bone marrow metastases, petechiae or purpura may occur due to thrombocytopenia. Neurological examination is also important in patients with CNS involvement. Initial examinations include an x-ray of the affected area; may show the “onion skin” appearance of periosteal reaction on x-ray. The primary site and potential metastatic areas should be evaluated with imaging tests. Plain radiographs of the affected area may show “moth-eaten” lesions that destroy bone, the “Codman triangle,” or a multilayered “onion skin” periosteal reaction. According to the updated 2017 National Comprehensive Cancer Network (NCCN) guideline, imaging of primary areas is done with MRI and, if necessary, CT, and MRI with contrast material administration is of primary importance. Other imaging modalities include chest CT, positron emission tomography (PET)/CT, and spine/pelvis MRI. The symptomatic patient should be referred to an orthopedic tumor surgeon if a biopsy is required. Diagnosis is preferably made by needle biopsy or open biopsy. Molecular cytogenetic analysis of biopsy samples should be included in the diagnostic workup to evaluate T(11;22) translocation. Bone marrow aspiration and bone marrow biopsy may be considered. According to NCCN guidelines, initial evaluation should include serum lactate dehydrogenase (LDH) as it has prognostic significance. Patients should also be offered fertility counseling (sperm egg freezing) before starting treatment.
Ewing Sarcoma Chemotherapy And Radiotherapy: Historically, Intergroup Ewing Sarcoma studies (IESS-I and IESS-II) compared RT+VACA (vincristine, dactinomycin, and cyclophosphamide) with VAC (vincristine, dactinomycin, and cyclophosphamide). showed better results with cyclophosphamide and doxorubicin). Due to doxorubicin dose limitation in dactinomycin regimens, subsequent studies did not show a significant effect on clinical outcomes when dactinomycin was excluded. Several studies have evaluated the addition of ifosfamide and etoposide to standard chemotherapy. Pediatric Oncology Group-Child Cancer Group study INT0091 showed that the VACD-IE group had significantly better survival rates than the VACD group.
In addition, the incidence of local failure was lower in the VACD-IE group. In the EICESS-92 study (European Intergroup Cooperative Ewing Sarcoma Study), VACA (vincristine, dactinomycin, cyclophosphamide and doxorubicin) and VAIA (vincristine, dactinomycin, ifosfamide and doxorubicin) were compared in standard risk patients (SR) and the effect of cyclophosphamide was found to be similar to ifosfamide; however, cyclophosphamide has been associated with increased toxicity. The 3-year event-free survival (EFS) rates were 73% and 74% for VACA and VAIA, respectively. The Euro-EWING99-R1 study (equivalence study based on the EICESS-92 protocol) evaluated whether cyclophosphamide could replace ifosfamide in consolidation therapy including vincristine and dactinomycin in standard-risk patients and compared VAC (vincristine, dactinomycin and cyclophosphamide) to VAI (vincristine, dactinomycin and cyclophosphamide). ifosfamide) suggested that it was not statistically inferior; however, VAI was associated with slightly higher 3-year event-free survival EFS. In a phase III study (AEWS0031) from the Children's Oncology Group (COG), patients received VDC alternating with IE every three weeks. The study showed that 2-week intervals were more effective than 3-week intervals without increasing toxicity.
These results led to VDC/IE becoming the standard of care in the United States. Chemotherapy is started before local treatment and continued after surgery if there are no signs of progression. Surgical resection and RT are local control treatment approaches. To date, there are no studies comparing the effectiveness of these two approaches. The INT-0091 study found no significant differences in local failure or event-free survival between surgery alone and RT alone. However, surgery plus RT has been found to be associated with a lower incidence of local failure. Despite this, we believe that disease recurrence is inevitable in cases where only RT is given without surgical treatment. Data from 1058 patients from the CESS 81, CESS 86 and EICES-92 studies showed that surgery+RT and surgery alone had a significantly lower local failure rate than RT alone. The incidence of local failure was similar in the preoperative RT group and the surgical group with or without postoperative RT. Based on data obtained from patients enrolled in studies INT-0091, INT-0154 or AEWS0031, it was shown that surgery + RT was associated with a lower risk of local failure compared to RT alone.
2. Chondrosarcoma
Chondrosarcoma Treatment: Chondrosarcoma Symptoms:
What is Chondrosarcoma?
Chondrosarcoma is a type of bone cancer that develops from cartilage cells. Cartilage is specialized connective tissue found in adults and is the tissue from which most bones develop. Cartilage plays an important role in the growth process. There are many different types of cartilage in the body. Chondrosarcoma primarily affects cartilage cells in the femur (thigh bone), arm, pelvis, or knee. Other areas (such as the ribs) may also be affected, although less frequently. Chondrosarcoma is the second most common type of primary bone cancer. Primary bone cancer is cancer that starts in the bone. This type of cancer rarely affects individuals under the age of 20. The risk continues to increase until age 75. The incidence is equal between men and women.
What Causes Chondrosarcoma?
The exact cause of chondrosarcoma is unknown. There may be a genetic or chromosomal component that makes some individuals more susceptible to such malignancies. Chondrosarcomas have been observed as a late consequence of radiation therapy for other cancers.
What are the Risk Factors for Chondrosarcoma?
Most often, chondrosarcoma arises from normal cartilage cells. It may also result from a pre-existing benign (non-cancerous) bone or cartilage tumor. Below is a list of some benign conditions that may be present when chondrosarcoma occurs:
- Enchondromas. A type of benign bone tumor that starts in the cartilage and usually affects the hands (it can also affect other areas).
- Multiple exostoses (osteochondromas). Presence of multiple osteochondromas
- Ollier's disease. A group of enchondromas (benign cartilage tumors that usually affect the hands).
- Maffucci syndrome. A combination of multiple enchondromas (benign cartilage tumors that usually affect the hands) and angiomas (benign tumors consisting of blood vessels).
What are the symptoms of chondrosarcoma?
Chondrosarcoma symptoms may vary depending on the location of the tumor. The following are the most common symptoms of chondrosarcoma. However, symptoms may occur differently in each individual. Symptoms may include:
- Large mass in the affected bone
- Feeling of pressure around the mass
- Pain that gradually increases over time. It usually gets worse at night and can be relieved by taking anti-inflammatory medications such as ibuprofen. It is usually not relieved by rest.
- Local swelling
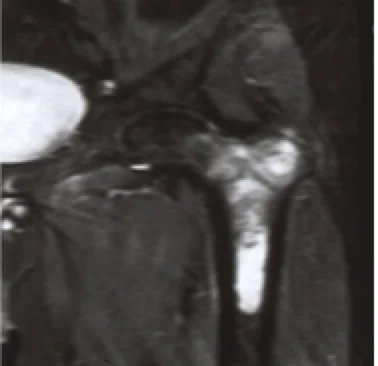
How is Chondrosarcoma Diagnosed?
In addition to a complete medical history and physical examination, diagnostic procedures for chondrosarcoma may include the following:
- X-ray. It is a diagnostic test that uses invisible beams of electromagnetic energy to convert images of internal tissues, bones, and organs into film.
- Computed tomography scan (also called CT or CAT scan). This is an imaging test that uses X-rays and a computer to create detailed images of the body. A CT scan shows details of bones, muscles, fat and organs.
- Magnetic resonance imaging (MRI). A diagnostic procedure that uses a combination of large magnets, radiofrequencies, and computers to take detailed images of organs and structures in the body.
- Positron emission tomography (PET) scan. An imaging test in which radioactively labeled glucose (sugar) is injected into the bloodstream. Tissues that use glucose more than normal tissues (such as tumors) can be detected by a scanning machine.
- Biopsy. A procedure in which tissue samples are removed from the body (either by injection or during surgery) for examination under a microscope. This is done to determine if cancer or other abnormal cells are present.
Chondrosarcoma Treatment:
Specific treatment for chondrosarcoma will be determined by your healthcare provider based on:
- Your age, general health and medical history
- Type, stage (spread) and location of cancer
- Your tolerance to certain medications, procedures, and therapies
- Expectation regarding the course of the disease
- Your opinion or preference
The goal of chondrosarcoma treatment is to remove the mass and reduce the likelihood of it returning. Treatment may include:
- Surgery. Removal of the tumor. If the tumor is in an arm or leg, the surgeon will try to save the limb. In some cases, amputation may be needed.
- Physical Therapy. This treatment helps regain strength and use of the affected area after surgery.
- Radiation therapy. Radiation can be given in high doses.
- Chemotherapy. Although it is not a primary treatment, it may be needed if the cancer has spread to other parts of the body.
3. Osteosarcoma
What is osteosarcoma?
Osteosarcoma (OS) or osteogenic sarcoma (OGS) (or bone cancer for short) is a cancerous tumor in the bone. Specifically, it is an aggressive malignant neoplasm arising from primitive transformed cells of mesenchymal origin, exhibiting osteoblastic differentiation and producing malignant osteoid. Osteosarcoma is the most common histological form of primary bone sarcoma. It is most common in teenagers and young adults.
Osteosarcoma Symptoms:
Many patients first complain of long-standing pain that worsens at night, can be intermittent and of variable intensity. Young people who are actively involved in sports often complain of pain in the lower part of the thigh bone or just below the knee. If the tumor is large, it may present as a prominent localized swelling. Sometimes a sudden fracture may be the first symptom because the affected bone is not as strong as normal bone and can break abnormally with minor trauma. In deeper-seated tumors that are not very close to the skin, such as those originating from the pelvis, localized swelling may not be evident.
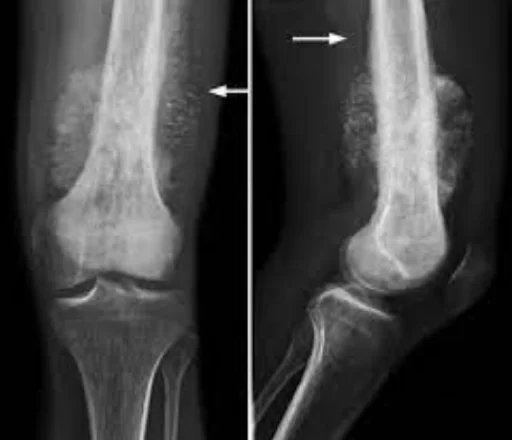
Osteosarcoma Causes:
Familial cases in which deletion of chromosome 13q14 inactivates the retinoblastoma gene are associated with a high risk of developing osteosarcoma. Bone dysplasias, including Paget's disease of bone, fibrous dysplasia, enchondromatosis, and hereditary multiple exostoses, increase the risk of osteosarcoma.
Li-Fraumeni syndrome (germline TP53 mutation) is a predisposing factor for the development of osteosarcoma. Rothmund-Thomson syndrome (i.e., autosomal recessive association of congenital bone defects, hair and skin dysplasias, hypogonadism, and cataracts) is associated with an increased risk of this disease. High doses of Sr-90 increase the risk of bone cancer and leukemia in animals and are predicted to do the same in humans.
Does Fluoride Cause Osteosarcoma?
There is no clear relationship between water fluoridation and cancer or cancer-related deaths, both for cancer in general and bone cancer and osteosarcoma in particular. A number of studies have concluded that fluoride concentration in water is not associated with osteosarcoma. Beliefs regarding the relationship between fluoride exposure and osteosarcoma stem from a 1990 study from the US National Toxicology program that showed equivocal evidence of a relationship between fluoride and osteosarcoma in male rats. However, there is still no solid evidence that fluoride has a tendency to cause cancer in mice. Fluoridation of water is practiced worldwide to improve the dental health of citizens. It is also considered a great health achievement. Fluoride concentration levels in water supplies are regulated, such as the United States Environmental Protection Agency regulating fluoride levels to be no more than 4 milligrams per liter. In fact, naturally occurring fluoride already exists in water supplies, but many communities have chosen to add more fluoride to the point that it can reduce tooth decay. Fluoride is also known for its ability to cause new bone formation. However, further research shows that fluoridated water does not pose a risk of osteosarcoma in humans. Most studies involved counting the number of osteosarcoma patients in specific areas with different concentrations of fluoride in drinking water. Statistical analysis of the data shows that there is no significant difference in the incidence of osteosarcoma cases in different fluoridated regions. Another important study involved collecting bone samples from osteosarcoma patients to measure fluoride concentration and compare them with bone samples of newly diagnosed malignant bone tumors. The conclusion is that mean fluoride concentrations in bone samples of osteosarcoma patients and tumor controls were not significantly different. It has been proven that not only the fluoride concentration in bones, but also the fluoride exposure of osteosarcoma patients does not differ significantly from healthy people.
Osteosarcoma tends to occur in areas of bone growth; This is probably because proliferation predisposes osteoblastic cells in this region to acquire mutations that could lead to transformation of the cells (RB gene and p53 gene). The tumor may be located at the end of the long bone (usually in the metaphysis). It most often affects the proximal end of the tibia or humerus or the distal end of the femur. Osteosarcoma tends to affect areas around the knee in 60% of cases, around the hip in 15%, the shoulder in 10%, and the jaw in 8%. The tumor is solid, hard, and irregular because the tumor spicules in the calcified bone radiate at right angles (“fir tree,” “moth-eaten,” or “sunset” appearance on x-ray examination). These right angles form what is known as the Codman triangle; this is a characteristic but not diagnostic feature of osteosarcoma.
Osteosarcoma Diagnosis:
X-rays are the preferred initial imaging method to diagnose osteosarcoma. Some features of osteosarcoma on x-rays are the sunset view and the Codman triangle (elevation of the bone cortex of the tumor causing new bone formation). CT scanning is useful in defining bone anatomy, the integrity of the bone cortex, detecting pathological fracture, and assessing ossification (laying of new bone materials) and cartilage calcification. On the other hand, soft tissue and medullary cavity are better visualized with MRI scanning.
A biopsy of suspected osteosarcoma should be performed by a qualified orthopedic oncologist. The American Cancer Society states: “Probably in no other case of cancer is it more important that this procedure be performed correctly. “An improperly performed biopsy can make it difficult to save the affected limb from amputation.” It can also metastasize to the lungs and appear mainly as single or multiple round nodules in the lower regions on chest X-ray.
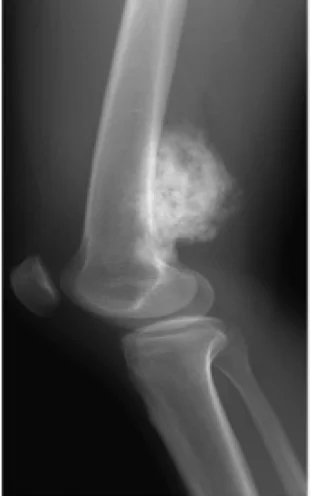
Osteosarcoma Types:
- Conventional: osteoblastic, chondroblastic, fibroblastic
- Telangiectatic
- Small cell
- Low grade central
- Periosteal
- Paraosteal
- Secondary
- High grade surface
- Extraskeletal
Osteosarcoma Treatment:
Complete surgical, en bloc resection of the cancer in osteosarcoma is the preferred treatment method. Although most patients can undergo limb salvage surgery, complications (especially infection, prosthesis loosening and nonunion, or local tumor recurrence) may lead to the need for further surgery or amputation.
PATIENTS WITH OSTEOSARCOMA ARE BEST MANAGED BY AN ORTHOPEDIC ONCOLOGIST EXPERIENCED IN SARCOMA TREATMENT. The current standard treatment is neoadjuvant chemotherapy (chemotherapy given before surgery) followed by surgical resection. The percentage of tumor cell necrosis (cell death) seen in the tumor after surgery gives an idea about the prognosis and also allows the oncologist to know whether the chemotherapy regimen needs to be changed after surgery.
Standard treatment is limb-saving orthopedic surgery (or amputation in some cases) when possible and leucovorin rescue with high-dose methotrexate, a combination of intra-arterial cisplatin, adriamycin, mesna with ifosfamide, BCD (bleomycin, cyclophosphamide, dactinomycin) etoposide and muramyl tripeptide. The protocol used is an aggressive intra-arterial regimen that individualizes treatment based on arteriographic response. In some studies, three-year event-free survival ranges from 50% to 75%, while five-year survival ranges from 60% to 85%. Overall, 65-70% of patients treated five years ago will be alive today. These survival rates are general averages and vary greatly depending on the individual necrosis rate.
Osteosarcoma Course of the Disease:
Grade I osteosarcoma is rare and includes parosteal osteosarcoma and low-grade central osteosarcoma. It has an excellent prognosis (>90%) with extensive resection. The overall prognosis depends on the location of the tumor (proximal tibia, femur, pelvis, etc.), the size of the tumor mass, and the degree of necrosis resulting from neoadjuvant chemotherapy. Other pathological factors, such as the degree of P-glycoprotein, whether the tumor is cxcr4-positive or Her2-positive, are also important because these are associated with distant metastases to the lung. Mortality rates due to osteosarcoma are decreasing by approximately 1.3% per year. Long-term survival probabilities for osteosarcoma improved significantly in the late 20th century, reaching approximately 68% in 2009.
