




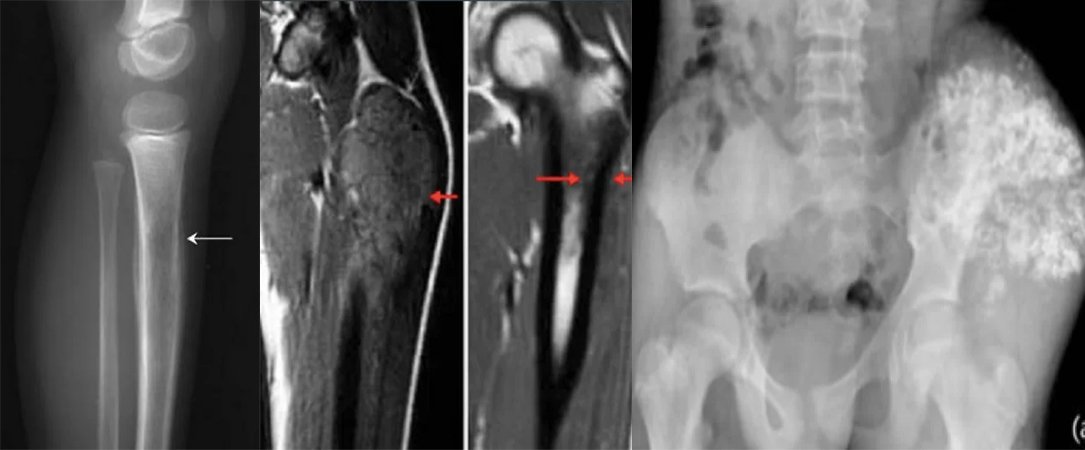
Sarcoma - Bone Cancer - Soft Tissue Cancer General Information
Bone Cancer-Bone Tumor: Primary bone tumor-bone cancer is a rare type of cancer that starts in the bones. In the United Kingdom (population 67 million people), 550 people are diagnosed with bone tumor-bone cancer every year (8 people per 1 million).
Bone Tumor Symptoms-Bone Cancer Symptoms: Although bone cancer can affect any bone, it usually affects the long bones in the arms and legs. Main symptoms:
- Continuous bone pain that increases in severity over time and continues at night
- Swelling and redness (limiting movement if the joint is close)
- Noticeable lump on bone
- Weak bone that breaks more easily than normal
- Limming
If you or your child has persistent, severe, or worsening bone pain, you should see an orthopedic oncologist.
What is a bone tumor?: Primary bone tumor-bone cancer is a rare type of cancer that starts in the bones. In the United Kingdom (population 67 million people), 550 people are diagnosed with bone tumor-bone cancer every year (8 people per 1 million).
Bone Tumor Surgery Recovery Time: What to Expect After Surgery?
Every surgery is different. You should ask your surgeon and treatment team what to expect immediately after surgery. In most cases you will encounter:
You will wake up in a recovery room. You will be monitored closely by healthcare providers. Once you are awake and stable, the staff will move you to a regular hospital room.
You will have a large bandage that will reduce your ability to move one of your limbs or joints. This will remain in place for a few days to a few weeks.
Your plastic drainage tubes may be stuck. The nurse empties these tubes. They will be removed after a few days when the drainage stops.
You will have pain and you will use painkillers. Medicines are usually given intravenously. Additionally, PCA (patient-controlled analgesia) can also be used, which you can control by pressing a button.
You will receive fluids through a small, flexible tube inserted into a vein in your arm. You will need this until you can eat and drink on your own. Most of the time, you will be able to eat within the 6th hour after surgery.
To prevent infection, you will receive antibiotics the day of surgery and sometimes for a day or two after surgery.
If necessary, a urinary catheter can be inserted for a few days. This is a tube that drains your urine.
After surgery, you may need chemotherapy or radiation to reduce the chance of remaining cancer cells growing and spreading. Applying another treatment method after surgery is called adjuvant treatment. The surgical incision must heal before you can begin this type of treatment. Adjuvant therapy can usually begin a few weeks after surgery.
Recovery time after surgery is different for each person. It depends on the type of surgery performed. Most people stay in the hospital for about two days to a week.
The wound will take about two weeks to heal, but it may take a month or two for your bone to heal completely. You may need physical therapy. Non-viable implants and internal prostheses can be inserted to fill the massive gap created after the tumor bone is removed, but these may result in infection, wound opening and loosening in the long term. To prevent these, we perform living bone transplants, which are more difficult and take longer to heal. Because an inanimate metal can never replace the bone tissue of a living and patient. In cases where living bone transplantation is performed, bone union and full weight bearing may take up to 1 year. However, this is still a long-term method that we always prefer, as your own bone is reconstructed and the gap is filled with living tissue, compared to non-living prostheses and implants. However, in some patients, internal prostheses may be preferred to living bone transplants. It is important to perform rehabilitation after limb-sparing surgery. This will help you learn how to use your arm or leg and make sure it is working well.
If you have had a limb-sparing surgery, other surgeries may be needed if any complications develop. In children, as the other leg or arm grows, more surgery may be needed to lengthen the limb. This ensures that both limbs remain the same length.
Bone Tumor Treatment: We use a combination of surgery, radiation therapy, chemotherapy, targeted therapies (smart drugs, tyrosine kinase inhibitors, etc.) in the treatment of bone tumors. Your treatment plan is created according to the type of cancer, which bone and area it is in, its stage and other factors.
Smart Drug: Targeted Therapy: As researchers better understood the intracellular gene changes that cause bone cancer, they developed new drugs that target these changes. These drugs work by a different mechanism than chemotherapy drugs, and their side effects are different from chemotherapy. They are often effective in bone tumors where chemotherapy does not work well (e.g. chordoma). Other non-chemotherapy drugs are bone-directed drugs and immunotherapies. Targeted drugs are called kinase inhibitors. Kinases are intracellular signals that indicate that cells should grow. Blocking kinases slows or stops the growth of some tumors. While these drugs are often used in the treatment of chordoma, some can also be used in advanced chondrosarcomas. Examples of kinase inhibitors:
- Imatinib
- Dasatinib
- Sunitinib
- Erlotinib
- Lapatinib
- Sorafenib
- Regorafenib
- Pazopanib
These medications come in the form of pills taken once or twice a day. Side effects include symptoms such as nausea, muscle pain and fatigue.
Denosumab is a RANKL inhibitor. Although it has been recommended for use in the treatment of giant cell tumors by some authorities in recent years, recent studies have reported that it is effective in the transformation of giant cell tumors into malignant tumors, and therefore it has lost its popularity. Interferon alfa-2b may be recommended in recurrent giant cell tumor or metastases of giant cell tumor. Side effects of this treatment include flu-like muscle aches, bone pain, fever, headache, fatigue, nausea and vomiting.
What is Sarcoma: Sarcoma Symptoms: What is Bone Cancer:
Sarcoma is a malignant tumor, a type of cancer arising from cells of mesenchymal (connective tissue) origin. Connective tissue is a broad term that includes bone, cartilage, fat, vascular or other structural tissues, and sarcomas can occur in any of these tissue types. As a result, there are many subtypes of sarcoma, classified according to the specific tissue and cell type from which the tumor originates. Sarcomas are primary connective tissue tumors, meaning they arise from connective tissues. This is different from secondary (or “metastatic”) connective tissue tumors, which occur when a cancer elsewhere in the body (such as the lungs, breast tissue, or prostate) spreads to connective tissue. Sarcomas are one of five different types of cancer, classified according to the type of cell from which they arise. The word sarcoma is derived from the Greek σάρκωμα sarōma 'fleshy protrusion or substance', itself from σάρξ sarx meaning 'flesh'.
Sarcoma Classification: Sarcoma Types: Sarcoma Types:
Sarcomas are typically divided into two main groups: bone sarcomas and soft tissue sarcomas. Each of these has multiple subspecies. In the United States, the American Joint Committee on Cancer (AJCC) publishes guidelines classifying sarcoma subtypes. These subtypes are as follows:
Subtypes of Bone Sarcoma
- Osteosarcoma
- Chondrosarcoma
- Poorly differentiated round/spindle cell tumors (including Ewing sarcoma)
- Hemangioendothelioma
- Angiosarcoma
- Fibrosarcoma/myofibrosarcoma
- Chordoma
- Adamantima
- Other:
- Liposarcoma
- Leiomyosarcoma
- Malignant peripheral nerve sheath tumor
- Rhabdomyosarcoma
- Synovial sarcoma
- Malignant solitary fibrous tumor.
Subtypes of Soft Tissue Sarcoma: Soft Tissue Cancer: What is Soft Tissue Cancer?
- Liposarcoma (Liposarcoma Cancer) (includes the following types: atypical lipomatous tumor/well-differentiated liposarcoma, undifferentiated liposarcoma, myxoid liposarcoma, pleomorphic liposarcoma, and myxoid pleomorphic liposarcoma)
- Atypical lipomatous tumor
- Dermatofibrosarcoma protuberans (includes pigmented varieties)
- Dermatofibrosarcoma protuberans, fibrosarcomatous
- Giant cell fibroblastoma
- Malignant solitary fibrous tumor
- Inflammatory myofibroblastic tumor
- Low grade myofibroblastic sarcoma
- Fibrosarcoma (includes adult and sclerosing epithelioid varieties)
- Myxofibrosarcoma (formerly myxoid malignant fibrous histiocytoma)
- Low grade fibromyxoid sarcoma
- Giant cell tumor of soft tissues
- Leiomyosarcoma
- Malignant glomus tumor
- Rhabdomyosarcoma (includes: embryonal, alveolar, pleomorphic, and spindle cell/sclerosing)
- Hemangioendothelioma (includes: retiform, pseudomyogenic, and epithelioid)
- Soft tissue angiosarcoma
- Extraskeletal osteosarcoma
- Gastrointestinal stromal tumor, malignant (GIST)
- Malignant peripheral nerve sheath tumor (includes epithelioid variety)
- Malignant Triton tumor
- Malignant granular cell tumor
- Malignant ossifying fibromyxoid tumor
- Stromal sarcoma unless otherwise stated
- Myoepithelial carcinoma
- Malignant phosphaturic mesenchymal tumor
- Synovial sarcoma (includes the following types: spindle cell, biphasic, and not otherwise specified)
- Epithelioid sarcoma
- Alveolar soft part sarcoma
- Clear cell sarcoma in soft tissue
- Extraskeletal myxoid chondrosarcoma
- Extraskeletal Ewing sarcoma
- Interdigitating dendritic cell sarcoma
- Desmoplastic small round cell tumor
- Extrarenal rhabdoid tumor
- Perivascular epithelioid cell tumor, not otherwise specified
- Intimal sarcoma
- Undifferentiated spindle cell sarcoma
- Undifferentiated pleomorphic sarcoma
- Undifferentiated round cell sarcoma
- Undifferentiated epithelioid sarcoma
- Undifferentiated sarcoma unless otherwise stated
Sarcoma Signs and Signs: Symptoms of Bone Tumor: Symptoms of Bone Cancer: Symptoms of Liposarcoma:
Symptoms of bone sarcomas typically include bone pain, especially at night, and swelling around the tumor site.
Symptoms of Soft Tissue Cancer in the Leg: Symptoms of Soft Tissue Cancer:
The symptoms of soft tissue sarcomas vary, but they usually present as hard, often painless lumps or nodules. Gastrointestinal stromal tumors (GIST, a subtype of soft tissue sarcoma) are often asymptomatic but may be associated with vague complaints such as abdominal pain, bleeding in the intestine, a feeling of fullness, or other signs of intestinal obstruction.
Sarcoma Causes and Risk Factors:
The cause of most bone sarcomas is unknown, but several factors are associated with an increased risk of developing bone sarcoma. Previous exposure to ionizing radiation (such as prior radiation therapy) is one of these risk factors. Therapeutic radiation is associated with sarcoma after 10 to 20 years. Exposure to alkylating agents found in some cancer chemotherapeutic drugs also increases the risk of bone sarcoma. Certain inherited genetic syndromes, such as Li-Fraumeni syndrome, hereditary RB1 gene mutations, and Paget bone disease, are associated with an increased risk of developing bone sarcoma. Most soft tissue sarcomas are caused by what doctors call “sporadic” (or random) genetic mutations in the affected person's cells. However, there are some risk factors associated with an increased risk of developing soft tissue sarcoma. Previous exposure to ionizing radiation is one such risk factor. Exposure to vinyl chloride (such as fumes encountered in polyvinyl chloride (PVC) production), arsenic, and Thorotrast are all associated with an increased risk of angiosarcoma.[2][3] Lymphedema resulting from certain types of breast cancer treatment is also a risk factor for developing angiosarcoma. As with bone sarcomas, certain inherited genetic syndromes, including Li-Fraumeni syndrome, familial adenomatous polyposis, neurofibromatosis type 1, and hereditary RB1 gene mutations, are also associated with an increased risk of developing soft tissue sarcoma. Kaposi's sarcoma is caused by Kaposi's sarcoma-associated herpes virus (HHV-8).
Sarcoma Mechanisms:
The exact molecular changes that result in sarcoma are not always known, but certain types of sarcoma are associated with certain genetic mutations. Examples include: Most cases of Ewing sarcoma are associated with a chromosomal translocation in which part of chromosome 11 is fused with part of chromosome 22. This causes the EWSR1 gene to fuse with other genes, including the FLI1 gene in 90% of Ewing cases and the ERG gene in 5-10% of cases. These fusions cause abnormal proteins to be produced, but it is not known exactly how these abnormal proteins lead to cancer. Dermatofibrosarcoma protuberans is often associated with a chromosomal translocation in which the COL1A1 gene is fused to the PDGFRB gene. This results in overactive PDGF signaling, which is thought to promote cell division and ultimately lead to tumor development. Inflammatory myofibroblastic tumor is often associated with rearrangement of the ALK gene and sometimes with rearrangement of the HMGA2 gene. Tenosynovial giant cell tumor (not a sarcoma but a non-metastasizing and locally aggressive soft tissue tumor) is often associated with a chromosomal translocation between chromosome 1 and chromosome 2, where the CSF1 gene joins the COL6A3 gene. This causes increased production of the CSF1 protein, which is thought to play a role in cancer development. Many liposarcomas are associated with an amplification of part of chromosome 12, resulting in extra copies of known cancer-promoting genes (“oncogenes”), such as the CDK4 gene, MDM2 gene, and HMGA2 gene.
Sarcoma Diagnosis: Bone Sarcomas:
Diagnosis of bone sarcomas begins with a detailed history and physical examination, which can reveal characteristic signs and symptoms (see Signs and Symptoms above). Some bone sarcomas (such as osteosarcoma) may be associated with elevated alkaline phosphatase levels, some bone sarcomas (such as Ewing sarcoma) may be associated with an elevated erythrocyte sedimentation rate, but laboratory studies are not particularly useful in diagnosis. Importantly, however, none of these laboratory findings are specific to bone sarcomas; This means that elevations in these laboratory values are associated with many other conditions besides sarcoma and therefore cannot be relied upon for a definitive diagnosis of sarcoma. Imaging studies are critical in diagnosis, and most clinicians will initially order plain radiography (x-rays). Other imaging studies commonly used in diagnosis include magnetic resonance imaging (MRI) studies and radioisotope bone scans. CT scanning is not typically used to diagnose most types of bone sarcomas, but it is an important tool for staging (see below). Definitive diagnosis requires biopsy of the tumor and careful examination of the biopsy sample by an experienced pathologist.
Soft Tissue Sarcomas: Soft Tissue Sarcoma: Soft Tissue Cancer: What is a Soft Tissue Tumor?
Diagnosis of soft tissue sarcomas begins with a detailed history and physical examination. Imaging studies may include CT or MRI, but CT tends to be preferred for soft tissue sarcomas located in the thorax, abdomen, or retroperitoneum. Positron emission tomography (PET) can also be useful in diagnosis, but its most common use is for staging purposes (see below). As with bone sarcomas, definitive diagnosis requires biopsy of the tumor with evaluation of histology by a trained pathologist.
Sarcoma Staging:
In general, cancer staging refers to how advanced the cancer is and is usually based on factors such as the size of the tumor and whether it has spread to other parts of the body. Staging is important because stage affects prognosis (likely outcome) as well as the types of treatments that are likely to be effective against the cancer. Staging for sarcomas involves determining whether the tumor has grown into surrounding tissues (“local invasion”), as well as whether it has spread to the lymph nodes (forming “nodal metastases”) or spread to other tissues or organs in the body (forming “distant metastases”). The most common imaging tools used to stage bone sarcomas are MRI or CT to evaluate the primary tumor, contrast-enhanced chest CT to assess whether the cancer has spread (i.e., metastasized) to the lungs, and radioisotope bone scan to assess whether it has spread to other bones. Staging for soft tissue sarcomas typically includes MRI or CT imaging of the primary tumor to determine tumor size, as well as contrast-enhanced chest CT to evaluate metastatic tumors in the lungs.
Grade - Sarcoma Degree:
Like some other cancers, sarcomas are given a grade (low, medium, or high) based on the appearance of the tumor cells under a microscope. In general, grade refers to how aggressive the cancer is and how likely it is to spread (“metastasize”) to other parts of the body. Low-grade sarcomas have a better prognosis than high-grade sarcomas and are usually treated surgically, although radiation therapy or chemotherapy is sometimes used. Intermediate and high-grade sarcomas are more often treated with a combination of surgery, chemotherapy, or radiotherapy. Because higher-grade tumors are more likely to metastasize (invade and spread to local and distant sites), they are treated more aggressively. The realization that many sarcomas are sensitive to chemotherapy has significantly increased the survival rate of patients. For example, before chemotherapy, the long-term survival of pediatric patients with localized osteosarcoma was only around 20%, but now this rate has increased to 60-70%.
Sarcoma Treatment: Bone Tumor Surgery: Bone Cancer Treatment: Liposarcoma Treatment:
What is Liposarcoma: Sarcoma Cancer: Soft Tissue Tumor: Soft Tissue Sarcomas:
Surgery is the most common treatment for most sarcomas that have not spread to other parts of the body, and surgery is the only curative treatment for most sarcomas. Limb-sparing surgery, as opposed to amputation, is now used in at least 90% of extremity (arm or leg) sarcoma cases to save patients' limbs. Additional treatments, such as chemotherapy including proton therapy, radiation therapy (also called “radiotherapy”), may be administered before surgery (called “neoadjuvant” chemotherapy or radiotherapy) or after surgery (called “adjuvant” chemotherapy or radiotherapy). The use of neoadjuvant or adjuvant chemotherapy and radiotherapy significantly improves the prognosis of many sarcoma patients. Treatment can be a long and difficult process, taking about a year for many patients. Liposarcoma treatment usually consists of surgical resection, and chemotherapy is considered depending on the aggressiveness of the sarcoma. Radiotherapy can also be used before or after surgical excision for liposarcoma. Pediatric rhabdomyosarcoma is usually treated with chemotherapy, surgery, and sometimes radiotherapy. The long-term survival rate in pediatric rhabdomyosarcoma patients is 50-85%. Osteosarcoma is a bone cancer that is treated by surgical removal of the cancer, usually combined with chemotherapy. Radiotherapy is a helpful alternative, although not as successful as surgery. In old years, it was believed that higher doses of chemotherapy could increase survival. However, high doses of chemotherapy stop the production of blood cells in the bone marrow and can be harmful. Stem cells collected from people before high-dose chemotherapy can be transplanted back into the person if their blood cell count becomes too low; This is called autologous hematopoietic stem cell transplantation or high-dose treatment with stem cell rescue. To investigate whether autologous hematopoietic stem cell transplantation following high-dose chemotherapy was more favorable than standard-dose chemotherapy, no significant difference was detected.
Sarcoma Life Time Prognosis Course:
The AJCC has identified several factors that affect the prognosis of bone sarcomas:
- Tumor size: Larger tumors tend to have a worse prognosis than smaller tumors.
- Tumor spread to surrounding tissues: Tumors that spread locally to surrounding tissues tend to have a worse prognosis than tumors that have not spread beyond their site of origin.
- Stage and presence of metastasis: Tumors that have spread (“metastasized”) to lymph nodes (rare for bone sarcomas) or other organs or tissues (for example, the lungs) have a worse prognosis than tumors that have not spread.
- Tumor grade: High-grade tumors (grade 2 and 3) tend to have a worse prognosis than low-grade (grade 1) tumors.
- Skeletal location: Tumors originating from the spine or pelvic bones tend to have a poorer prognosis compared to tumors originating from the bones of the arms or legs.
Factors affecting prognosis in soft tissue sarcomas other than GIST are:
- Stage: As with bone sarcomas, tumors that have metastasized have a worse prognosis than tumors that have not metastasized.
- Grade: The AJCC recommends the use of a grading system for soft tissue sarcomas called the French Federation of Cancer Centers Sarcoma Group (FNCLCC) Grade; High-grade tumors have a worse prognosis than low-grade tumors.
The main factor affecting the prognosis for GISTs:
- Mitotic rate: mitotic rate refers to the fraction of cells that are actively dividing within the tumor; GISTs with a high mitotic rate have a worse prognosis than GISTs with a low mitotic rate.
Sarcoma Survival:
According to data published by the US National Cancer Institute (NCI), the 5-year overall survival in bone sarcomas is 66.9%. The American Cancer Society (ACS) estimates that 2,140 people in the United States will die from bone sarcomas in 2023, accounting for 0.3% of all cancer deaths. The average age of death is 61, but death can occur in any age group. Accordingly, 12.3% of deaths from bone sarcoma occur in people under the age of 20, 13.8% in people between the ages of 20-34, 5.5% in people between the ages of 35-44, and 9.3% in people over 45 years of age. -In people aged 54, 13.5% in people aged 55-64, 16.2% in people aged 65-74, 16.4% in people aged 75-84 and 13.1% It is seen in people aged 85 and over. The overall 5-year survival for soft tissue sarcomas (regardless of stage) is 64.5%, but survival is affected by many factors, including stage. Thus, 5-year survival is 80.8% for soft tissue sarcomas that have not spread beyond the primary tumor (“localized” tumors), 58.0% for soft tissue sarcomas that have spread only to nearby lymph nodes, and 16.4% for soft tissue sarcomas that have spread to distant organs. type. The ACS estimates that 5,140 people will die from soft tissue sarcoma in 2023, accounting for 0.9% of all cancer deaths.
Epidemiology:
Sarcomas are rare cancers. The risk of being diagnosed with a new bone cancer in a previously healthy person is less than 0.001%, while the risk of being diagnosed with a new soft tissue sarcoma is between 0.0014 and 0.005%. The American Cancer Society estimates that 3,970 new cases of bone sarcoma and 13,400 new cases of soft tissue sarcoma will occur in the United States in 2023. Given that the estimated total number of new cancer diagnoses (all cancer types) is 1,958,310, this suggests that bone sarcomas represent only 0.2% of all new cancer diagnoses (making them the 30th most common type of cancer) and soft tissue shows that sarcomas represent only 0.7% of all new cancer diagnoses (making them the 22nd most common type of cancer). Sarcomas affect people of all ages. Approximately 50% of bone sarcomas and 20% of soft tissue sarcomas are diagnosed in people under 35 years of age. Some sarcomas, such as leiomyosarcoma, chondrosarcoma, and gastrointestinal stromal tumor (GIST), are more common in adults than in children. Most high-grade bone sarcomas, including Ewing sarcoma and osteosarcoma, are much more common in children and young adults. In 2016, scientists reported that an osteosarcoma tumor was discovered in a 1.6-1.8 million-year-old fossil found in the skeleton of the extinct hominin species Australopithecus sediba, making it the oldest known case of human cancer. So bone cancer and other cancers have been affecting hominins and humans for about 2 million years. The steps in diagnosis and treatment only cover a period of 100 years.
Bone Tumor Surgery Prices: Bone tumor surgery prices vary depending on the type of tumor, its location, the number of specialists who will perform the surgery, the extent of damage, and whether limb-saving microsurgery procedures are performed or not.
Tumor Treatment in the Leg: Tumors in the leg are removed by preserving the vessels and nerves or by transplantation when necessary.
